

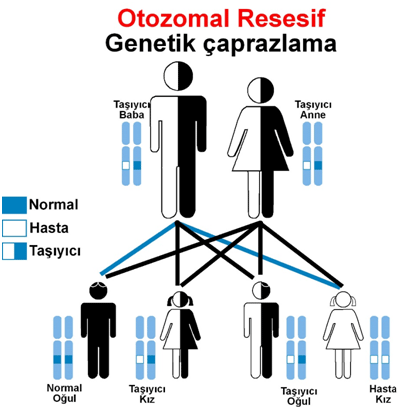
Anne ve babadan genetik olarak geçen metabolik bir hastalıktır. Çekinik genle taşınan bu hastalığın taşıyıcı sıklığı ülkemizde yüksektir. Her 100 kişiden dördünün bu hastalığı taşıyor olması yanı sıra %22 ‘ye varan akraba evliliği sıklığı hastalığın ülkemizde sık görülmesinin nedenidir.
Türkiye fenilketonüri hastalığının en sık görüldüğü ülkeler arasındadır. Doğan her 6.000 – 8.000 çocuktan biri fenilketonüri hastası olarak dünyaya gelmektedir.
Fenilketonüri hastalığının genetik aktarımının şeması:

Ülkemiz hastalığın en sık izlendiği ülkelerdendir. Doğması beklenen bebek sayısı ile değerlendirildiğinde her yıl 200-300 yeni fenilketonüri vakasının topluma katılacağı hesaplanmaktadır.
Hastalıkta protein yapıtaşı olan fenilalanin adlı aminoasidi karaciğerde tirozin adlı aminoaside dönüştüren fenilalanin hidroksilaz enziminin normal çalışmaması sonucu kanda fenilalanin birikimi ile meydana gelir.

Kanda biriken fenilalanin ve geriye dönüşümsüz beyin hasarına neden olur. Erken tanımlanıp tedavi edilmediği takdirde kaçınılmaz son ağır zihinsel geriliktir.
Ağır zihinsel geriliği olan fenilketonürili bireylerde konvülsiyonlar, agresif yada otistik davranış bozuklukları, dermatit şeklindeki cilt lezyonları yanı sıra vakaların % 60’ında anne babaya göre açık saç-göz-ten rengi ile karekterize görünüm vardır.
Fenilketonüri hastalığının kandaki fenilalanin düzeyine göre 2 farklı tipi vardır:
1.Klasik Fenilketonüri: Kanda bakılan fenilalanin düzeyi 20 mg/dl (1200 µmol/L) üzerinde olan hastalar bu gruba girer. Bu hastalarda fenilalanin hidroksilaz enzimi hiç çalışmamaktadır.
Klasik Fenilketonüri hastaları tanı konulduğundan itibaren fenilalaninden kısıtlı diyet yapmaları gerekmektedir.
Diyet tedavisi ömür boyudur.
Fenilalanin tüm proteinli gıdaların içinde bulunmaktadır (kırımızı/beyaz et, süt, peynir, yoğurt, yumurta, ekmek…). Bu hastalar, içinde fenilalanin bulunmayan özel ürünler kullanmaları ve vegan diyet (bitkisel gıda ağırlıklı) gerekmektedir.
Diyete erken yaşta başlamak ve uymak zihinsel etkilenmenin önlenmesinde en önemli faktördür.
Hastaların yaşına ve fenilalanin değerlerine göre belli aralıklarla kan fenilalanin düzeyine bakılmalı, diyetleri metabolizma uzmanları ve diyetisyenler ile ayarlanmalıdır. Kanda bakılan fenilalanin düzeyinin 2-6mg/dl (120-360 µmol/L) arasında tutulması ağır zeka etkilenmesini engellemektedir.
2. Hiperfenilalaninemi: Bu grup kendi içinde iki alt gruba ayrılmaktadır
a. Hafif Hiperfenilalaninemi: Normal diyetle izlemde kan fenilalanin düzeyi 2-10 mg/dl (120-600 µmol/L) arasında seyreder.
Bu hastalarda fenilalanin düzeyi 2-6 mg/dl (120-360 µmol/L) arasında olanlarda diyet tedavisine gerek yoktur. Fakat olası fenilalanin düzeyi yükselmelerine karşı rutin Metabolizma Bölümünde takibi gerekmektedir.
b. Hiperfenilalaninemi: Kan fenilalanin düzeyi 10-20 mg/dl arasında olan hastalardır.
Kan fenilalanin düzeyinin 6mg/dl üzerine çıkması durumunda zeka etkilenebileceği için bu grup hastalar da fenilalaninden kısıtlı diyet yapmaları veya ilaç tedavileri almaları gerekmektedir. Bu gruptaki olgular Klasik Fenilketonürili hastalara göre diyet tedavilerindeki kısıtlamalar genellikle daha hafif olmaktadır.
Fenilketonüri hastalarına diyet tedavisi ne kadar erken başlanırsa zeka etkilenmesi o kadar az olur. Bu sebepten dolayı hastalık ülkemizde topuk kanı taraması programına alınmıştır.
Malign Fenilketonüri (Tetrahidrobiopterin metabolizması bozuklukları):
Fenilalanin hidroksilaz enziminin çalışmasında gerekli olan tetrahidrobiyopterin’in (BH4) geri dönüşüm bozukluğuna bağlı ortaya çıkan durum “Malign Fenilketonüri” olarak adlandırılmaktadır.
BH4 sadece Fenilalanin Hidroksilaz enziminin değil ayrıca Tirozin Hidroksilaz ve Triptofan Hidroksilaz enziminin de normal çalışması için gereklidir. Bu enzimlerinde çalışamamasına bağlı olarak sinir sisteminin normal çalışması için gerekli olan seratonin ve dopamin sentezi yapılamaz.
Bu olgularda BH4 yanında L-dopa/carbidopa ve 5-OH-triptofan desteği de yapılması gerekmektedir. Bazı olgular fenilalaninden kısıtlı diyete de ihtiyaç duyarlar. Nörolojik etkilenme bu olgularda klasik fenilketonüriye göre daha ağırdır.

Fenilketonüri Hastalarının İzlemi:
Hasta izlemleri kan fenilalanin düzeyi ile yapılmaktadır.
Bebeklerde daha sıkı takip önerilirken yaş arttıkça takip aralıkları da uzamaktadır. Her ülkenin kendisine göre bir takip planı mevcut olmakla birlikte merkezler arası takip planları değişiklik gösterebilmektedir.
Amaç kan düzeyini bebeklerde 2-6 mg/dl (120-360 µmol/L) arasında tutmak iken
12 yaş üstü hastalarda 2- 10 mg/dl (120-600 µmol/L) arası olması önerilir.
Her ne kadar takip aralıkları ülkeden ülkeye değişse de her hasta kendi içinde değerlendirilmelidir.
Tedavinin hedefi diyet ile yeterli büyümenin sağlanması, kan fenilalanin artışına bağlı oluşan nörolojik etkilenmenin ve beslenme kısıtlamalarına nedeni ile oluşabilecek vitamin, mineral ve eser element eksiklerinin önüne geçilmesini sağlamaktır.
Tedavi ömür boyudur.
Erişkin hastalarda diyete uyumsuzluk sonucu kanda yükselen fenilalanin seviyelerinin dikkat eksikliğine, agresif davranışlara, arka arkaya hızlı şekilde yapılan işlemlerde yavaşlama gibi sinirsel ve psikiyatrik sorunlara yol açtığı ve diyete uyumlu ile kan fenilalanin seviyesi düştüğünde bu bulguların ortadan kalktığı gösterilmiştir.
Fenilketonüri hastalarının hayvansal gıda tüketememelerinden dolayı hastalarda B12 vitamini, folik asit, demir, çinko, bakır, magnezyum, fosfat, selenyum, kalsiyum, esansiyel yağ asitleri (çoklu doymamış yağ asitleri) eksiklikleri görülebilmektedir. Bu durumun önüne geçebilmek için, hastalara vitamin ve mineral desteği verilmeli, vitamin mineral eksiklikler açısından belli aralıklarla tetkik edilmeli ve büyüme ve gelişmeleri takip edilmelidir.
Kemik mineral yoğunluğu, bu olgularda nedeni tam olarak açıklanamayan bir sebepten dolayı kendi yaş gruplarına göre geri olabilmektedir. Fenilketonürili hastalarda kemik erimesi (osteoporoz, osteopeni) yönünden değerlendirilmeli ve kalsiyum D vitamini desteği verilmelidir.
Belli aralıklarla rutin nöropsikiatrik testlerle etkilenmeleri ortaya konulmalıdır.
Diyet tedavisi ömür boyudur. Zamanla diyet tedavisinde esneklikler yapılsa bile yükselen kan fenilalanin düzeyi olgularda nöropsikiatrik etkilenmelere neden olmaktadır.
Fenilketonüri Tedavisinde Yenilikler:
Enzim yerine koyma tedavisi: Fenilalanin liyaz enzimi memeli canlılarda olmayan bitkilerde bulunan bir enzimdir. Fenilalanin’i toksik olmayan maddelere dönüşmesini sağlar.

Bu enzim cilt altına iğne ile verilmesi uygulanmaktadır. Dünyada ve ülkemizde uzun dönem tedavi sonuçları araştırılmaktadır.
Large nötral aminoasit içeren tablet ve toz ürünler: Large nötral aminoasitleri (LNAA) (L-Tyrosine, L-Tryptophan, L-Methionine, L-Threonine, L-Isoleucine, L-Valine, L-Leucine, L-Histidine, L-Lysine, L-Arginine, Phe bir LNAA’dır) içeren bir üründür. Bu aminoasitler kandaki fenilalanin beyne geçişini ve bağırsaklardan fenilalanin emilimini engelleyerek sinir sistemini korumaktadır. İlaç yemeklerle birlikte alınmaktadır.
Özellikle erişkin yaş hastalarda kullanılmaktadır. Yüksek fenilalaninin yol açtığı dikkat eksikliğine, agresif davranışlara, seri yapılan işlemlerde yavaşlama gibi sinirsel ve psikiyatrik durumlara olumlu etkileri olduğu belirtilmektedir.
LNAA içeren ürünler Türk Eczacılar Birliğinden tedarik edilmektedir.

Sapropterin (Kuvan, Diterin): Sapropterin fenilalanin hidroksilaz enziminin çalışmasında yardımcı olan bir maddedir. Fenilalanin hidroksilaz enziminin kısmı eksikliğinde mevcut enzimin dahaz hızlı çalışmasını sağlar. Klasik PKU hastalarının %30’unun ve hafif PKU hastaların %80’inin bu tedaviden fayda gördüğü belirtilmektedir.
Hastaların sapropterine yanıt verip vermeyeceği saptropterin yükleme testi ile yapılmaktadır: sapropterin tablet formu ağızdan verildiğinde kan fenilalanin düzeyinde %30 düşüş saptanması klinik olarak yanıt verdiğini göstermektedir.
Ayrıca genetik tahlil ile de hastanın tedaviden fayda görebileceği belirlenebilir.

Karaciğer nakli: Nakil sonrası oluşabilecek sorunlar düşünüldüğünde önerilmeyen bir yöntemdir.
Gen Tedavisi: Şu an yapılamamaktadır. Klinik çalışmalar devam etmektedir.
Fenilketonürili hastalar için ülkemizde çeşitli dernekler bulunmaktadır.
Bu derneklerde aileler bir araya gelmekte ve çeşitli etkinlikler düzenlenmektedir.
Ayrıca dernek ve Metvak Vakfı aracılığı ile hastaların tüketebileceği özel ürünler tedarik edilebilmektedir.


Fenilketonüri hastalığı tanı, tedavi ve takibi için merkezimize başvurabilirsiniz. Merkez bilgilerimiz için tıklayınız.
FENİLKETONÜRİ HAKKINDA GÜNCEL YAZIMIZA BURAYA TIKLAYARAK ULAŞABİLİRSİNİZ
Bu konu hakkında bize soru sorabilirisiniz.












Yorum yazabilmek için oturum açmalısınız.