

Biotinidaz Eksikliği Nedir?
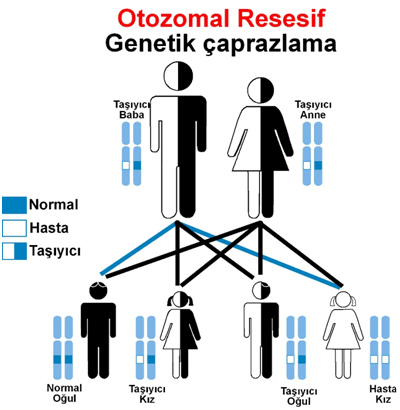
Biotinidaz eksikliği, anne ve babadan genetik olarak geçen (otozomal resesif) bir metabolik hastalıktır.
Dünya genelinde nadir görülmekle birlikte, biyotinidaz eksikliğinin en sık görüldüğü ülkelerden birinin Türkiye olduğu bildirilmiştir. Ülkemizde yenidoğanlarda görülme sıklığı yaklaşık 1:1100 olarak rapor edilmiştir
(Baykal T, Hüner G, Sarbat G, et al. Acta Paediatr, 1998).
Biotin ve Biotinidazın Görevi
Biotin (Vitamin B7, Vitamin H), vücutta dört farklı karboksilaz enziminin normal çalışması için gerekli olan önemli bir vitamindir.
Biotinidaz enzimi ise, “biotin döngüsü” adı verilen süreçte biyotinin tekrar kullanılmasını sağlar. Bu sayede vücut biyotin ihtiyacını etkin şekilde karşılayabilir.
Biotinidaz eksikliğinde bu döngü bozulur ve vücutta serbest biyotin düzeyi azalır. Sonuç olarak birçok metabolik süreç etkilenir.
Biotinidaz Eksikliğinde Görülen Bulgular
Tedavi edilmediğinde hastalık şu bulgularla ortaya çıkabilir:
- Metabolik asidoz (kanda asit oranının artması)
- Deri bulguları: saç dökülmesi, egzema, cilt lezyonları
- Nörolojik belirtiler: nöbetler, kas güçsüzlüğü, gelişme geriliği
- İşitme ve görme problemleri
Geç kalınan vakalarda koma ve ölüm görülebilir.
Yenidoğan Tarama Programı
Bu ciddi sonuçlar nedeniyle biotinidaz eksikliği 2007 yılından bu yana Türkiye’de yenidoğan tarama programı kapsamında, bebeklerden alınan topuk kanı ile taranmaktadır.
Tarama sayesinde hastalık henüz belirtiler ortaya çıkmadan saptanabilmektedir.
Tedavi ve İzlem
Biotinidaz eksikliğinin tedavisi oldukça basit ve etkilidir:
- Biotin vitamini ağızdan verilir
(genellikle 5–20 mg/gün) - Tedavi ömür boyu sürdürülmelidir
Enfeksiyon, ateş veya cerrahi girişim gibi vücudun metabolizma hızının arttığı durumlarda biyotin dozu geçici olarak artırılabilir.
🔹 Verilen biotin tedavisi enzim aktivitesini düzeltmez, ancak biyotin eksikliğine bağlı bulguların ortaya çıkmasını önler.
Tedavinin Etkisi
- Deri ve nörolojik bulgular tedavi ile genellikle düzelir
- Tedavi öncesinde gelişmiş işitme ve görme kayıpları ise çoğu zaman kalıcı olabilir
Bu nedenle yenidoğan taraması ile tanı alan bebeklerde, vakit kaybetmeden biotin tedavisine başlanması hayati öneme sahiptir.
Takip
Biotinidaz eksikliği tanısı alan hastalar:
- Düzenli aralıklarla izlenmeli
- İşitme ve göz muayeneleri açısından takip edilmelidir
Biotinidaz Eksikliği – Sık Sorulan Sorular
Biotinidaz eksikliği geçici bir hastalık mıdır?
Hayır. Biotinidaz eksikliği genetik geçişli ve ömür boyu süren bir hastalıktır. Bu nedenle tedavisi de ömür boyu devam eder.
Biotinidaz eksikliğinin kesin tanısı nasıl konur?
Hastalık öncelikle biotinidaz enzim aktivitesinin ölçülmesi ile saptanır. Türkiye’de yenidoğan tarama programında da bu yöntem kullanılmaktadır.
Kesin tanı için ise genetik analiz yapılması gereklidir.
Biotinidaz eksikliği gebelikte yaşanan sorunlara (enfeksiyon, beslenme vb.) bağlı mıdır?
Hayır. Biotinidaz eksikliği anne ve babadan genetik olarak geçen bir hastalıktır. Gebelik sırasında yaşanan enfeksiyonlar, kullanılan ilaçlar veya beslenme şekli hastalığa neden olmaz.
Aynı hastada farklı zamanlarda bakılan biotinidaz enzim aktivitesi değişebilir mi?
Evet. Enzim aktivitesi ölçümü birçok faktörden etkilenebilir.
Bunlar arasında:
- Numunenin uygun koşullarda alınmaması veya saklanmaması
- Laboratuvar tekniği farklılıkları
- Yenidoğanda sarılık
- Kullanılan bazı antibiyotikler
yer almaktadır. Bu nedenle enzim aktivitesi zaman zaman farklı sonuçlar verebilir.
Biotin tedavisinin yan etkisi var mıdır?
Hayır. Biotin suda çözünen bir vitamindir. Fazlası idrarla atılır ve bilinen ciddi bir yan etkisi yoktur.
Biotin tedavisi ile biotinidaz enzim aktivitesi düzelir mi?
Hayır. Biotin tedavisinin amacı, biotinidaz enzimi çalışmadığı için gelişen biyotin vitamini eksikliğini yerine koymaktır.
Verilen biotin, enzim aktivitesini artırmaz veya düzeltmez.
Biotinidaz eksikliği olan bireylerin günlük yaşamda dikkat etmesi gereken özel durumlar var mıdır?
Hayır. Hastalar biotin tedavisini düzenli kullandıkları ve takiplerini aksatmadıkları sürece, sağlıklı bireyler gibi normal yaşamlarını sürdürebilirler.
Her türlü besini tüketebilir, gerekli olduğunda diğer tıbbi tedavileri güvenle alabilirler.
📌 Ancak enfeksiyon, ateş veya ameliyat gibi metabolizmanın hızlandığı durumlarda, geçici olarak biotin dozunun artırılması, biyotin eksikliğinin önlenmesi açısından faydalı olabilir.
Biotin tedavisi düzenli kullanılsa bile kontroller gerekli midir?
Evet. Tedavi düzenli kullanılsa bile hastalar:
- Nörolojik muayene
- Gelişimsel değerlendirme
- Göz ve işitme muayeneleri
açısından belirli aralıklarla takip edilmelidir. Gerekirse biotin dozu bu değerlendirmelere göre yeniden düzenlenebilir.
Biotin tedavisi kan tahlillerini etkiler mi?
Evet. Biotin kullanımı sırasında bazı laboratuvar testlerinde yanlış düşük veya yanlış yüksek sonuçlar görülebilir.
Özellikle:
- 25-OH D vitamini
- Parathormon (PTH)
- Tiroid fonksiyon testleri
biotin kullanımından etkilenebilir. Bu nedenle hekimlerin ve ailelerin bu konuda farkındalığı, yanlış tanı ve tedavilerin önlenmesi açısından çok önemlidir.
Ailede biotinidaz eksikliği tanısı alan biri varsa diğer bireyler taranmalı mıdır?
Evet. Özellikle yenidoğan taraması ile tanı alan vakalarda, anne, baba ve kardeşlerin biotinidaz eksikliği açısından değerlendirilmesi önemlidir.
Ülkemizde tarama programının görece yeni olması nedeniyle, tarama öncesi doğmuş ve belirtisiz bireyler aile içinde bulunabilir. Klinik deneyimler, bu bireylerde tanı konulmasının ve semptom olmasa bile tedavi başlanmasının koruyucu olduğunu göstermektedir.
Biotinidaz eksikliği tanı, tedavi ve takibi için nereye başvurabilirim?
Biotinidaz eksikliği tanı, tedavi ve takip için merkezimize başvurabilirsiniz. Merkez bilgilerimiz için tıklayınız.
Bu konu hakkında bize soru sorabilirisiniz.















Yorum yazabilmek için oturum açmalısınız.