Son yazılar

Biotinidaz Eksikliği Nedir? Tanı, Tedavi ve Merak Edilenler
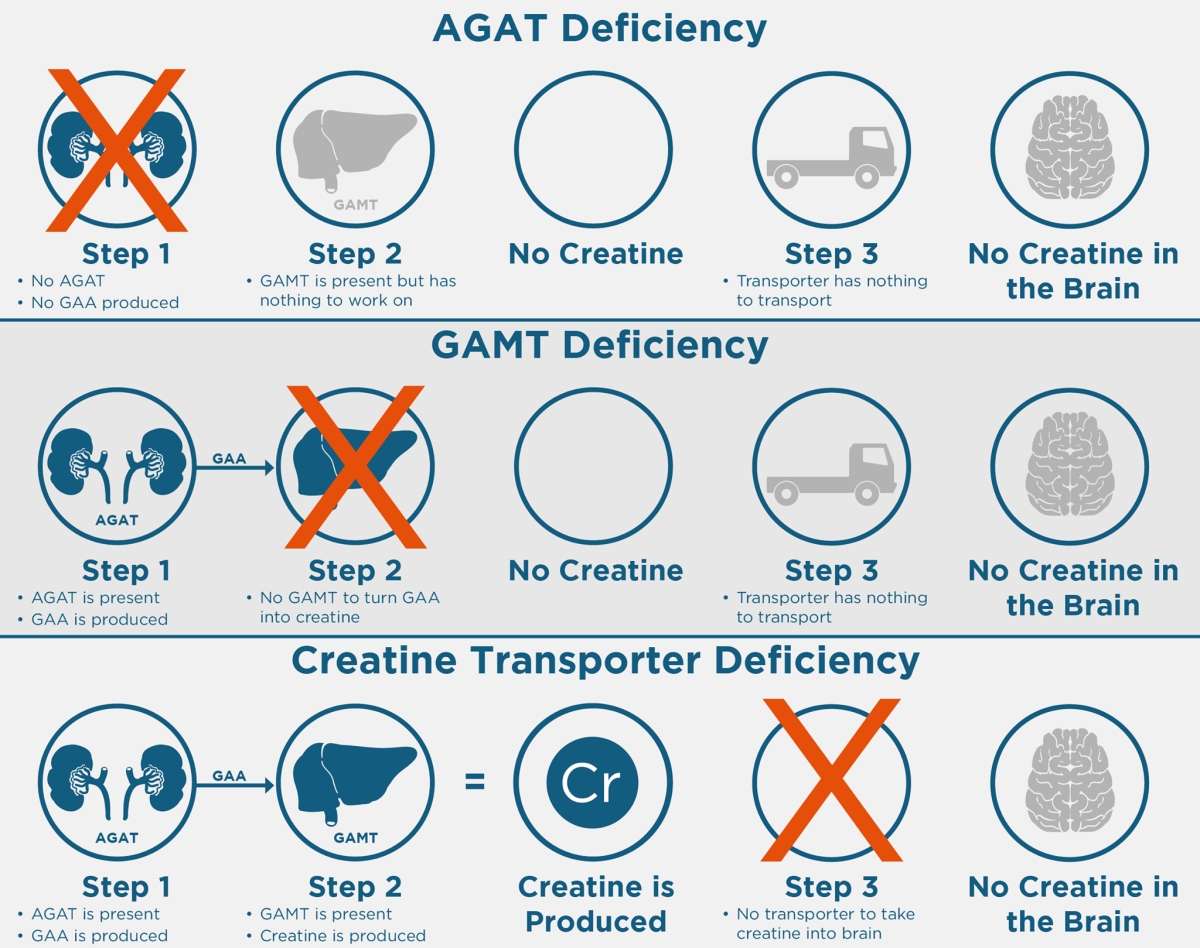
Biotinidaz Eksikliği Nedir? Biotinidaz eksikliği, anne ve babadan genetik olarak geçen (otozomal resesif) bir metabolik hastalıktır. Dünya genelinde nadir görülmekle birlikte, biyotinidaz eksikliğinin en sık görüldüğü ülkelerden birinin Türkiye olduğu bildirilmiştir. Ülkemizde yenidoğanlarda görülme sıklığı yaklaşık 1:1100 olarak rapor edilmiştir(Baykal T, Hüner G, Sarbat G, et al. Acta Paediatr, 1998). Biotin ve Biotinidazın Görevi Biotin…

Yenidoğan Taraması: Topuk Kanı Testinde Bilinmesi Gereken 7 Kritik Nokta
Yenidoğan tarama programı, bazı ciddi ve nadir hastalıkların belirti vermeden önce saptanmasını sağlayan, yaşam boyu sonuçları olan çok önemli bir halk sağlığı uygulamasıdır. Doğru zamanda ve doğru şekilde yapılan tarama, geri dönüşü olmayan hasarların önlenmesini sağlar. 1) Yenidoğan Tarama Programı Nedir? Türkiye’de Sağlık Bakanlığı tarafından tüm yenidoğan bebeklere uygulanan Ulusal Yenidoğan Tarama Programı, bazı metabolik…

EMZİRME DÖNEMİNDE ANNLERİN KULLANMAMASI GEREKEN İLAÇLAR
Kategori Açıklama İlaçlar / Gruplar Kontrendike (Kullanılmamalı) Bu ilaçlar emzirme döneminde kullanılmamalıdır. Eğer anne sağlığı için zorunluysa, emzirmeye geçici veya kalıcı olarak ara verilmelidir. Amantadin, Amiodaron, Antineoplastik ajanlar, Bromidler, Kloramfenikol, Kokain, Dipiron (Meksika ilaçlarında), Altın tuzları, Indandion antikoagülanlar, İyot (topikal formlar dahil), Metimazol, Metronidazol (emzirme geçici olarak durdurulmalı), Radyofarmasötikler (emzirme geçici olarak durdurulmalı), Salisilatlar (yüksek…


X’e Bağlı Adrenolökodistrofi (X-ALD)
X-ALD, beyinde miyelin (sinirleri saran koruyucu kılıf), adrenal bezler (böbreküstü bezleri) ve testislerde hasara neden olan nadir, X kromozomuna bağlı resesif kalıtsal bir peroksizomal bozukluktur. Hastalık, uzun zincirli yağ asitlerinin (VLCFA) parçalanmasını sağlayan bir taşıyıcı protein olan ABCD1 genindeki mutasyon sonucu oluşur. Kimleri Etkiler? Patofizyoloji ABCD1 genindeki mutasyon nedeniyle peroksizomlar, çok uzun zincirli yağ asitlerini…

SİTRİN EKSİKLİĞİ
Sitrin eksikliği, karaciğerin enerji üretimi ve protein işleme süreçlerinde görevli bir taşıyıcı proteinin (SLC25A13 geninde oluşan mutasyonlar nedeniyle) düzgün çalışmaması sonucu ortaya çıkan kalıtsal bir metabolik hastalıktır. Otozomal resesif geçer, yani anne ve babadan birer hatalı gen alınması gerekir. Hastalık genellikle üç farklı yaş döneminde kendini gösterir: 1. Yenidoğan Döneminde (NICCD)Sitrin eksikliği, yenidoğanlarda “Yenidoğana Özgü…

Lizinürik Protein İntoleransı (LPI)
Lizinürik Protein İntoleransı (LPI), vücudun bazı temel aminoasitleri (özellikle lisin, arginin ve ornitin) yeterince emip geri kazanamaması ile karakterize kalıtsal (genetik geçişli) bir metabolik hastalıktır. Bu aminoasitler proteinlerin yapı taşlarıdır ve sağlıklı büyüme, bağışıklık sistemi, kas yapısı ve üre döngüsü gibi birçok vücut işlevi için gereklidir. LPI, SLC7A7 adlı gendeki bir mutasyon nedeniyle ortaya çıkar…

KETON YAPIM (KETOGENEZ) KUSURLARI
Vücudumuz, enerji ihtiyacını karşılamak için farklı yollar kullanır. Keton yapım süreci (ketogenez), özellikle açlık dönemlerinde veya uzun süreli egzersiz sırasında beyin ve diğer organlar için önemli bir enerji kaynağı oluşturur. Ancak, bazı çocuklarda yağlardan keton üretiminde (ketogenez) genetik veya metabolik nedenlere bağlı olarak bozukluklar görülebilir. Bu durum, enerji üretiminde sorunlara yol açarak çeşitli sağlık problemlerine…

Ketoliz Defektleri
Ketoliz defektleri, vücudun yağlardan elde edilen keton cisimlerini enerji kaynağı olarak kullanamamasıyla karakterize edilen nadir kalıtsal metabolik hastalıklardır. Bu durum, özellikle açlık, uzun süreli egzersiz veya hastalık gibi enerji ihtiyacının ve yağ kullanımının arttığı açlık dönemlerinde belirgin hale gelir. Vücutta biriken keton cisimleri uygun şekilde metabolize edilemediğinden, hastalar enerji üretiminde ciddi sorunlar yaşayabilirler. Hastalığın Nedeni…

MPS III (Sanfilippo Sendromu)
MPS III (Sanfilippo Sendromu) Nedir? MPS III, Sanfilippo Sendromu olarak da bilinen nadir bir genetik hastalıktır. Lizozomal depo hastalıkları grubuna dahildir ve dört farklı alt tipi vardır (MPS IIIA, MPS IIIB, MPS IIIC, MPS IIID). Hastalık, vücutta glikozaminoglikan (GAG) adı verilen bir maddenin parçalanamaması ve birikmesi nedeniyle ortaya çıkar. Bu birikim özellikle merkezi sinir sistemini…

PROLİDAZ EKSİKLİĞİ
Prolidaz Eksikliği Nedir? Prolidaz eksikliği, vücudun bazı proteinleri parçalayamamasına neden olan nadir, kalıtsal bir metabolik hastalıktır. Özellikle kollajen gibi prolin açısından zengin proteinlerin yıkımı bozulur ve vücutta birikim meydana gelir. Bu durum, cilt, bağışıklık sistemi, akciğerler, kemikler ve sinir sisteminde çeşitli sağlık sorunlarına yol açabilir. Belirtileri Nelerdir? Prolidaz eksikliğine sahip bireylerde aşağıdaki belirtiler görülebilir: Hastalık…

Maroteaux-Lamy Sendromu (Mukopolisakkaridoz Tip VI, MPS VI)
Maroteaux-Lamy Sendromu, N-asetilgalaktozamin 4-sülfataz (Arylsulfataz B) enziminin eksikliği veya yokluğu nedeniyle ortaya çıkan nadir bir genetik hastalıktır. Bu enzim eksikliği, glikozaminoglikanlar (GAG) olarak bilinen kompleks şeker zincirlerinin hücre içindeki lizozomlarda birikmesine yol açar. Sonuç olarak, çeşitli organ ve dokularda ilerleyici hasarlar meydana gelir. Belirtiler: Hastalığın belirtileri, GAG birikiminin miktarına ve biriktiği dokulara bağlı olarak değişkenlik…

MPS Tip 4 (Morquio Sendromu)
Mukopolisakkaridoz Tip 4 (MPS Tip 4), nadir görülen genetik bir hastalıktır ve Morquio Sendromu olarak da bilinir. Bu hastalık, vücudun bazı karbonhidratları (glikozaminoglikanları – GAG’ları) parçalayamaması sonucu ortaya çıkar. Bu durum, özellikle iskelet sistemi ve bağ dokularında birikime neden olarak ilerleyici kemik bozukluklarına yol açar. MPS Tip 4’ün Nedenleri MPS Tip 4, vücuttaki belirli enzimlerin…

Mukopolisakkaridoz Tip 2 (MPS Tip 2)
Mukopolisakkaridoz Tip 2 (MPS Tip 2), lizozomal depo hastalıkları grubuna ait, nadir görülen bir genetik hastalıktır. Hunter Sendromu olarak da bilinir ve X’e bağlı resesif geçiş gösterir. Bu nedenle genellikle erkeklerde görülür ancak nadir de olsa kız vakalar bildirilmiştir. Hastalık, iduronat-2-sülfataz (I2S) enziminin eksikliği nedeniyle glikozaminoglikanların (GAG) hücrelerde birikmesi ile ortaya çıkar. Belirtileri Nelerdir? MPS…

Mukopolisakkaridoz Tip 1 (MPS Tip 1)
Mukopolisakkaridoz Tip 1 (MPS Tip 1), lizozomal depo hastalıkları grubuna ait, nadir görülen genetik bir hastalıktır. Vücudun belirli enzimleri yeterince üretememesi sonucu mukopolisakkarit adı verilen maddeler hücrelerde birikir ve organlara zarar verir. MPS Tip 1’in üç alt tipi bulunmaktadır: Belirtileri Nelerdir? MPS Tip 1 belirtileri, hastalığın şiddetine göre değişebilir. En yaygın görülen belirtiler şunlardır: MPS…

Ketojenik Diyet
Ketojenik diyet, düşük karbonhidrat, yüksek yağ ve orta düzeyde protein içeren bir diyet modelidir. Bu beslenme şekli, vücudu glikoz yerine yağları enerji kaynağı olarak kullanmaya yönlendirir. Yağların parçalanması sonucu oluşan keton cisimleri (asetoasetat, beta-hidroksibütirat ve aseton), özellikle beyin gibi glikoz bağımlı dokular için alternatif bir enerji kaynağıdır. Metabolik Süreçler: Epilepside Ketojenik Diyet: Mekanizma ve Etkileri…

Zellweger Spektrum Bozuklukları
Zellweger Spektrum Bozuklukları (ZSB), peroksizom adı verilen hücre içi organellerin oluşum ve işlev bozukluğuyla karakterize, nadir görülen genetik bir hastalık grubudur. Bu hastalıklar, peroksizomal biyogenez bozuklukları (PBD) olarak da adlandırılır ve genellikle ciddi nörolojik, karaciğer ve diğer organ problemleriyle ilişkilidir. Hastalığın Temel Özellikleri ZSB, genetik geçişli otozomal resesif hastalıklardır. Hastalık anneden ve babadan geçer. Peroksizomlar,…

AROMATİK L-AMİNO ASİT DEKARBOKSİLAZ (AADC) EKSİKLİĞİ
Aromatik L-Amino Asit Dekarboksilaz (AADC) eksikliği, çok nadir görülen genetik bir metabolizma bozukluğudur. Bu hastalıkta serotonin ve dopamin gibi önemli nörotransmitterlerin üretimi azalır. Sinir sistemi işlevleri için gerekli bu kimyasalların eksikliği, hareket, davranış ve vücut kontrolünde sorunlara yol açar. Hastalık, dünya genelinde 100.000 ila 500.000 doğumda 1 görülür ve oldukça nadirdir. AADC eksikliği, DDC (Dopa…

HİPERTRİGLİSERİDEMİ (TRİGLİSERİT YÜKSEKLİĞİ)
Hipertrigliseridemi, kandaki trigliserit seviyesinin yüksek olması durumudur. Bu durum, genetik ve çevresel faktörlerin bir kombinasyonuna bağlı olabilir ve pankreatit (pankreasın iltihaplanması) gibi ciddi sağlık sorunlarına yol açabilir. Nedenleri Belirtiler Hipertrigliseridemi genellikle belirti vermez; ancak trigliserit seviyeleri çok yüksek olduğunda şunlar görülebilir: Tip I Hiperlipoproteinemi (Şilomikronemi Sendromu) Genetik Nedenler Bu alt gruplar, farklı genetik hastalıklara dayalı…

GLİKOJEN DEPO HASTALIĞI TİP 1 (VON GİERKE HASTALIĞI)
Glikojen Depo Hastalığı Tip 1 (GDH Tip 1), çocukların enerji dengesini sağlamasını zorlaştıran nadir bir genetik hastalıktır. Bu hastalıkta karaciğer, böbrek ve bağırsak gibi organlarda enerji için gereken şekeri serbest bırakmayı sağlayan bazı enzimler düzgün çalışmaz. Bunun sonucunda kandaki şeker seviyesi çok düşebilir (hipoglisemi) ve bazı organlarda glikojen birikimi görülebilir. Hastalık Tipleri: GDH Tip 1a…



KLASİK GALAKTOZEMİ
Klasik galaktozemi, galaktoz-1-fosfat üretemeyen veya işleyemeyen bir enzim olan galaktoz-1-fosfat üridiltransferaz (GALT) eksikliği nedeniyle oluşan kalıtsal bir metabolik bozukluktur. Bu durum, galaktozun normal olarak parçalanmasını ve işlenmesini engeller. Klasik galaktozemi, otozomal resesif bir kalıtım deseni gösterir. Yani, hastalık için etkili bir genin her iki kopyasını taşıyan bireylerde belirtiler ortaya çıkar. Anne ve babanın her ikisi…

HEREDİTER FRUKTOZ İNTOLERANSI
Herediter fruktoz intoleransı (HFI), fruktoz adı verilen bir şekerin işlenmesini engelleyen kalıtsal bir bozukluktur. HFI, fruktoz intoleransı olarak da bilinir ve karaciğerdeki bir enzim olan fruktoz-1-fosfat aldolaz eksikliği nedeniyle ortaya çıkar. Bu eksiklik fruktozun etkili bir şekilde parçalanmasını engeller, fruktoz birikir ve toksik etkilere neden olur. Herediter fruktoz intoleransı (HFI), otozomal resesif bir kalıtım deseni…

AİLESEL HİPERKOLESTEROLEMİ
Ailesel hiperkolesterolemi (AH), kalıtsal bir lipid metabolizma bozukluğudur ve yüksek kolesterol düzeylerine yol açar. AH’nin temel nedeni, vücutta kolesterolü düzenleyen LDL reseptörlerinde bir bozukluk veya eksiklik olmasıdır. Bu durum LDL kolesterolünün yüksek seviyelerde birikmesine ve arterlerde plak oluşumuna yol açarak kalp hastalığı riskini artırır. AH, genetik bir bozukluktur ve genellikle otozomal dominant yolla aktarılır. Bu,…

ALFA MANNOSİDOZ
Alfa Mannosidoz, alfa-mannosidaz enziminin eksikliği ile karakterize nadir görülen bir genetik hastalıktır. Bu durum kompleks şeker moleküllerinin çeşitli organ ve dokularda birikmesine yol açarak çok çeşitli semptomlara yol açar. Alfa Mannosidoz, MAN2B1 genindeki mutasyonların neden olduğu otozomal resesif bir hastalıktır. Bir çocuğun etkilenmesi için her iki ebeveyn de mutasyona uğramış genin bir kopyasını taşımalıdır. Klinik…

BİOTİNİDAZ EKSİKLİĞİ VE SIK SORULAN SORULAR
Hastalık genetik geçiş göstermektedir ve ömür boyudur. Tedavi de ömür boyudur. Hastalık öncelikle biotinidaz enzim aktvitesi bakılarak teşhis edilir. Ülkemizde de enzim aktivitesi bakılarak yenidoğanlarda taranmaktadır. Kesin tanı için genetik analiz yapılmalıdır. Hayır. Hastalık hem anneden hem de babadan genetik olarak geçer. Gebelikte yaşanılan sorunlar biotinidaz eksikliğine neden olmaz. Evet. Biotinidaz enzim aktivitesi bir çok…

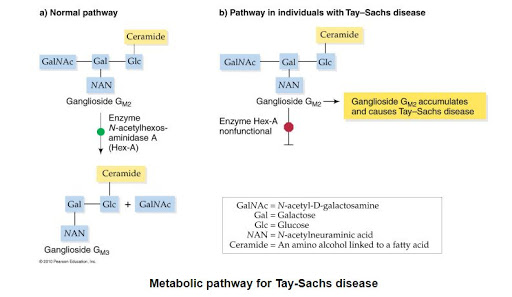
GM2 GANGLOİSİDOZ (TAY SACHS VE SANDHOFF)
3 farklı tipi mevcuttur. Tay-Sachs hastalığı (B varyant): HEXA geni 15. kromozomdaki mutasyona bağlı. Sandhoff hastalığı (0 varyant): HEXB genindeki mutasyona bağlı. GM2 aktivatör protein defekti (AB varyant): GM2 aktivatör protein genindeki (5.kromozomda) mutasyona bağlı ortaya çıkar. Hastalık hem anneden hem babadan otozomal resesif (çekinik) kalıtılır. TAY-SACHS beta-heksozaminidaz alfa- subuniti mutasyonu sonucu Heksozaminidaz A (alfa-beta…

GM1 GANGLİOSİDOZ
Bir tür lizozomal depo hastalığıdır. Hücre içindeki lizozom adı verilen organellerin içinde çeşitli maddelerin metabolize edilmemesi ve birikmesi sonucu ortaya çıkar. Otozomal resesif (çekinik) olarak kalıtılır. Hastalık hem anneden hem babadan geçiş gösterir. Hastalık bulgularının görülme yaşına göre klinik olarak farklılık göstermektedir. Tipik Erken İnfantil Bbebeklik) Form (Tip1): (enzim aktivitesi %0.1) Yaşamın ilk bir haftasında…

METAKROMATİK LÖKODİSTROFİ
Klinik: Metakromatik lökodistrofi (MLD) sinir sistemini tutan bir lizozomal depo hastalığıdır. Hastalık hem anneden hem babadan geçmektedir. Otozomal resesif (çekinik) olarak kalıtılmaktadır. Geç infantil form (Geç bebeklik formu): En sık görülen formudur. Bulgular 1-2 yaşında başlar. Olguların çoğu yürüyebilir. %15’i ise yürüyememektedir. Hastalar ilk bulgu olarak 14-16. Aylarda yürümede ve sıralama güçlük ve düşme şikayetleri…




HOMOSİSTİNÜRİ (KLASİK HOMOSİSTİNÜRİ)
Homosistinüri: İdrarda artmış homosistin atılımı anlamında olmakla birlikte esas olarak sistatiyonin β-sentetaz enzim eksikliğine bağlı gerçekleşen ‘’klasik homosistinüri’’yi tanımlamak için kullanılır. Bu hastalarda doğuştan enzim eksikliğine bağlı olarak vücutta homosisitein düzeyi artar. Hastalığın dünyada görülme 1:200.000-335000 arasındadır. Akraba evlilikleri tüm metabolik hastalıklarda olduğu gibi bu hastalığında görülme sıklığını arttırmaktadır. Hastalık anne ve babadan (otozomal resesif)…

GLUTARİK ASİDÜRİ TİP 1
Lizin ve Triptofan amino asitlerinin metabolizması bozukluğudur. Glutaril-KoA dehidrogenaz adlı verilen enzimin eksikliğine bağlı kan, idrar ve beyin omurilik sıvısında glutarik asit, 3-hidroksiglutrik asit ve glutarilkarnitin birikir. Biriken maddeler sinir sistemine olumsuz etkileri hastalığın klinik bulgularına neden olmaktadır. Hastalık anne ve babadan otozomal resesif genetik geçişlidir. KLİNİK: Hastaların doğum sonrası büyüme gelişmesi normal seyrederken (6ay-…

AKÇA AĞAÇ ŞURUBU İDRAR HASTALIĞI (MAPLE SYRUP URINE DISEASE) (MSUD HASTALIĞI)
Dallı zincirli aminoasit (Valin, Lösin, İzolösin) metabolizmasında kaynaklanan problem sonucu meydana gelir. Esas bulguların oluşmasında lösin ve metabolitlerinin birikimi sonucu görülür. Hastalık anne babadan otozomla resesif olarak kalıtılmaktadır. Akut tabloda valin, lösin, izolösin yanında artmış organik asitler (a ketoasitler) saptanır Dallı zincirli a ketoasit dehidrogenaz kompleks aktivitesinde sorun mevcuttur. Dallı zincirli a ketoasit dehidrogenaz kompleksin…

NİEMANN PİCK TİP C HASTALIĞI
Genetik, ilerleyici, geri dönüşümü olmayan, sinir sistemini ve diğer organları tutan bir lizozomal lipid depo hastalığıdır. Hastalı anne ve babadan genetik olarak geçiş göstermektedir. Kendine özgü bir fizik muayene, laboratuvar veya MR belirteci olmaması hastalığın tanısının konulmasında zorlaştırmaktadır. Erken tanı konulması durumunda nörolojik bulguların başında miglustat tedavisi başlanma şansı yakalanabilir. Erken tedavi bulguların önüne geçilmesinde…

TİROZİNEMİ TİP 2 (OKÜLOKUTANÖZ TİROZİNEMİ)
Tirozin aminotansferaz (TAT) enzim eksikliğine bağlı gelişir. Kromozom 16q22.1-q22.3 TAT gen mutasyonu bağlı ortaya çıkar. Hastalık anneden ve babadan otozomal resesif olarak kalıtılır. Palmoplantar hiperkeratoz denilen el içi ayak tabanında deri bulguları, gözde herpetiform korneal ülserler denilen karneal etkilenme ve zeka etkilenimi gözlenir. Karaciğer ve böbrek tutulumu olmaz. Göz bulguları ilk aylarda, cilt bulguları 1…

KRABBE HASTALIĞI (GLOBOİD HÜCRE LÖKODİSTROFİSİ)
Krabbe hastalığı merkezi sinir sistemini tutan doğuştan bir metabolik hastalıktır. Galaktoseramidi (sinir sisteminde bulunan myelin adı verilen sinir kılıfının yapısında yer alan bir madde) hücrelerde bulunan lizozom adı verilen yapılar içinde parçalanmasını sağlayan galaktoseramidaz (galaktoserebrosidaz, serebrozis b-galaktosidaz) enzim eksikliğine bağlı oluşur. Hastalık otozomal resesif olarak anneden ve babadan geçer. Galaktoseramidaz geni (GALC) 14q31 kromozomda yer…

SEREBROTENDİNÖZ KSANTOMATOZİS (STK)
STK her birinin başlangıcı ve ciddiyeti farklılık gösteren, hastalığa özgü bir dizi tabloya neden metabolik bir hastalıktır. STK nadir ve gerçekte olduğundan daha az tanı konulan otozomal resesif bir bozukluktur. Hastalık anne ve babadan otozomal resesif olarak geçer. STK’ya mitokondri enzimi olan “27-hidroksilaz”ı kodlayan CYP27A1 genindeki mutasyonlar neden olur. STK’da görülen ayırt ettirici tablolar arasında…

TİROZİNEMİ TİP 1 (HEPATORENAL TİROZİNEMİ)
Tirozinemi tip 1 bir tür amino asit metabolizması bozukluğudur. Tirozinemi tip 1 hastaları, yedikleri gıdalardan tirozin adı verilen bir amino asidi parçalamada sorun yaşarlar. Tedavi edilmezse, durum ciddi karaciğer hastalığına ve diğer ciddi sağlık sorunlarına neden olur. Hastalık anne ve babadan otozomal resesif şekilde genetik olarak geçer. Tirozinemi 1, fumarilasetoasetaz (FAH) adı verilen bir enzim…

NİEMANN-PİCK A/B (SFİNGOMYELİNAZ EKSİKLİĞİ)
Niemann Pick hücre içinde bulunan lizozom adı verilen yapıların içinde yer alan sfingomyelinaz adlı enzimin genetik olarak doğuştan eksikliği sonucu ortaya çıkan bir metabolik hastalıktır. Hastalıkta sfingomiyelinaz enzminin çalışamaması sonucu hücrelerde sfingomiyelin adlı maddenin birikimi meydana gelir. Bu birikim hastadaki şikayetlerin ve bulguların sebebidir. Hastalık anne ve babadan genetik olarak otozomal resesif şekilde kalıtılır. Hastalık…

PROPİYONİK ASİDEMİ
Besinlerle alınan ‘’Valin, metionin, treonin ve izolösin’’ adı verilen amino asitlerin ve yağ asitlerinin metabolize olmaması sonucu ortaya çıkan bir organik asidemidir. Anne ve babadan genetik (otozomal resesif) olarak geçer. Hastalar ‘’Valin, metionin, treonin ve izolösin’’ adı verilen amino asitlerin ve yağ asitlerini ihtiva eden besin ile beslendikten sonra metabolitleri olan propiyonik asit ve glisin…

METİLMALONİK ASİDEMİ (MMA)
Besinlerle alınan ‘’Valin, metionin, treonin ve izolösin’’ adı verilen amino asitlerin ve yağ asitlerinin metabolize olmaması sonucu ortaya çıkan bir organik asidemidir. Anne ve babadan genetik (otozomal resesif) olarak geçer. Hastalar ‘’Valin, metionin, treonin ve izolösin’’ adı verilen amino asitlerin ve yağ asitlerini ihtiva eden besin ile beslendikten sonra metabolitleri olan metilmalonik asit ve glisin…

İZOVALERİK ASİDEMİ
Besinlerle alınan ‘’lösin’’ adı verilen amino asidin metabolize olmaması sonucu ortaya çıkan bir organik asidemidir. Anne ve babadan genetik (otozomal resesif) olarak geçer (IVD geni). Bir çok metabolik hastalıkta olduğu gibi izovalerik asidemide de taşıyıcı anne ve taşıyıcı babanın sorunlu allellerinin çocuklarında yan yana gelmesi ile oluşur. Taşıyıcı anne babanın çocuklarının hasta olma ihtimali %25…
MATERNAL FENİLKETONÜRİ
Fenilketonüri tanılı annelerin gebelikleri sırasında, kandaki yüksek fenilalanin seviyesinin anne karnındaki bebeği etkilemesi durumuna maternal fenilketonüri denilmektedir. Bebekte doğum kilosunda düşüklük, baş çevresi küçüklüğü, anormal yüz bulgular, büyüme gelişme geriliği, göz ve kalp sorunları ve zeka geriliği görülebilir. Hafif hiperfenilalaninemili annelerde bu risk daha düşüktür. Fakat hiperfenilalaninemi tanılı annelerin de gebelikleri süresince mutlaka kan fenilalanin…

FABRY HASTALIĞI
Fabry Hastalığı X kromozumu üzerinden geçiş gösteren lizozomal depo hastalığıdır. Hastalık dünyada 40000-117000 kişide 1 görülmektedir. a-galaktosidaz A adlı enzim eksikliğine bağlı glikolipit birikimi [globotriaosilseramid (GL3)], farklı dokularda hücre içinde birikir. Enzim eksikliğinin derecesine göre klinik bulgularda değişiklik gösterebilmektedir. Klasik Fabry hastalığında; erken yaşlarda başlayan genellikle karın ve kasık bölgesindeki ciltte kırmızı kabarık döküntüler (anjiokeratoma…

POMPE HASTALIĞI
Hücrelerde bulunan lizozomlar adı verilen yapılar içinde yer alan asit α-glukozidaz enzim eksikliği nedeni ile ortaya çıkan genetik geçişli bir hastalıktır. Anne ve babadan genetik (otozomal resesif) olarak geçer (GAA geni). Klinik Belirtiler: Hastalık temelde iskelet kaslarını, kalp kasını ve düz kasları tutar. Hastalığın ağırlığı, başlangıç yaşına, organ tutulum derecesine ve kas tutulumunun ağırlığına bağlıdır.…

GAUCHER HASTALIĞI
Asit-b-glukosidaz enzim eksikliğine bağlı ortya çıkan hastalıktır. Glukoseramid adlı madde bu enzim ile glukoz ve seramide parçalanmaktadır. Enzim eksikliğinde ise vücudumuzun çeşitli yerlerindeki hücrelerinin içinde glukoseramid birikimi meydana gelir ve hastalık bulguları ortaya çıkar. Gaucher hastalığı GBA1 (Asit-b-glukosidaz) 1q21 genindeki mutasyon sonucu ortaya çıkar. Hastalık anne ve babadan (otozomal resesif) genetik olarak geçiş göstermektedir. Hastalıkta…

FENİLKETONÜRİ
Anne ve babadan genetik olarak geçen metabolik bir hastalıktır. Çekinik genle taşınan bu hastalığın taşıyıcı sıklığı ülkemizde yüksektir. Her 100 kişiden dördünün bu hastalığı taşıyor olması yanı sıra %22 ‘ye varan akraba evliliği sıklığı hastalığın ülkemizde sık görülmesinin nedenidir. Türkiye fenilketonüri hastalığının en sık görüldüğü ülkeler arasındadır. Doğan her 6.000 – 8.000 çocuktan biri fenilketonüri…

NON-KETOTİK HİPERGLİSİNEMİ
Karaciğerde bulunan “glisin cleavage” adı verilen enzim kompleksinin doğuştan eksikliği sonucu vücut sıvılarında (kan, beyin-omurilik sıvısı, idarda) glisin amino asidinin birikimi ile seyreden bir metabolik hastalıktır. Hastalık anne ve babadan genetik (otozomal resesif) olarak geçer. Bir çok metabolik hastalıkta olduğu gibi Nonketotik hiperglisinemi de taşıyıcı anne ve taşıyıcı babanın sorunlu allellerinin çocuklarında yan yana gelmesi…

ALKAPTONÜRİ
Homogentisat 1,2dioksijenaz enziminin doğuştan eksikliğine bağlı homogentisik asit’in (HGA) maleilasetoasetat’a dönüşememesi sonucu benzoquinon asetik asit (BQA), melanin- benzeri pigmentler birikimi ile seyreden bir hastalıktır. Homogentisat 1,2 dioksijenaz enzimi geni 3q13.33 kromozunda bulunmaktadır. Hastalık otozomal resesif olarak anneden ve babadan geçer. Otozomal resesif geçiş şeması: Metabolik hastalıkların çoğu otozomal resesif (çekinik kalıtım) şeklinde genetik olarak anne…