MPS III, Sanfilippo Sendromu olarak da bilinen nadir bir genetik hastalıktır. Lizozomal depo hastalıkları grubuna dahildir ve dört farklı alt tipi vardır (MPS IIIA, MPS IIIB, MPS IIIC, MPS IIID). Hastalık, vücutta glikozaminoglikan (GAG) adı verilen bir maddenin parçalanamaması ve birikmesi nedeniyle ortaya çıkar. Bu birikim özellikle merkezi sinir sistemini etkileyerek nörolojik bozukluklara neden olur.
Belirtileri Nelerdir?
İlk belirtiler (1-3 yaş): Konuşma ve motor gelişimde gecikme, sosyal iletişimde bozukluklar
İlerleyen dönem (3-10 yaş):
Dil ve motor gelişiminde gerileme
Hiperaktivite, agresif davranışlar, dikkat eksikliği
SGSH, NAGLU, HGSNAT ve GNS genlerinde patojenik mutasyonların tespiti ile kesin tanı konulabilir.
Tedavi Seçenekleri Şu anda MPS III için kesin bir tedavi bulunmamaktadır. Ancak hastalığın belirtilerini yönetmeye yönelik destekleyici tedaviler uygulanmaktadır:
Nörolojik ve Gelişimsel Destek:
Özel eğitim ve ergoterapi
Dil ve konuşma terapisi
Davranışsal destek programları
Farmakolojik Tedaviler:
Hiperaktivite ve davranış bozuklukları için risperidon gibi antipsikotik ilaçlar
Uyku problemleri için melatonin veya diğer uyku düzenleyici ilaçlar
Epilepsi varlığında uygun antiepileptik tedaviler
Fiziksel ve Ortopedik Destek:
Fizik tedavi ve egzersiz programları
Ortopedik destek (yürüme cihazları, oturma desteği)
Eklem sertliği ve skolyoz için takip
Solunum ve Kardiyovasküler Destek:
Tekrarlayan solunum yolu enfeksiyonları için düzenli takip ve tedavi
Oksijen terapisi veya solunum cihazı kullanımı
Kalp kapak hastalıkları için düzenli kardiyolojik takip
Beslenme ve Sindirim Desteği:
Yutma güçlüğü durumunda diyet düzenlemeleri
Beslenme tüpü (G-tüp) kullanımı ihtiyacı
Takip Edilmesi Gereken Sistemler ve Bölümler MPS III hastalarının düzenli olarak takip edilmesi gereken tıbbi bölümler şunlardır:
Nöroloji: Epilepsi, bilişsel gerileme ve hareket bozuklukları takibi
Genetik: Taşıyıcılık testleri ve genetik danışmanlık
Pediatri: Genel sağlık durumu ve büyüme gelişim takibi
Ortopedi: Eklem hareket kısıtlılığı, skolyoz ve kas-iskelet sistemi problemleri
Kardiyoloji: Kalp kapak hastalıkları ve ritim bozuklukları takibi
Fizik Tedavi ve Rehabilitasyon: Kas zayıflığı ve hareket kısıtlılıklarının yönetimi
Gastroenteroloji: Yutma güçlüğü, beslenme bozuklukları ve kabızlık takibi
Göğüs Hastalıkları: Solunum problemleri ve enfeksiyon risklerinin yönetimi
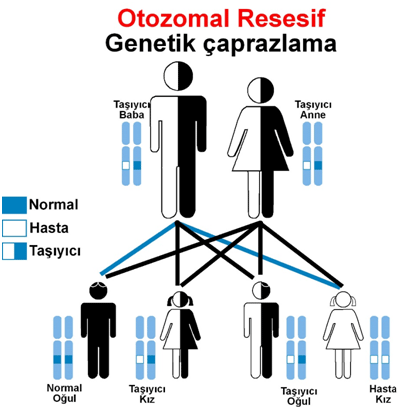
Genetik Danışmanlık ve Aile Desteği MPS III otozomal resesif bir hastalık olduğundan, ebeveynler taşıyıcıdır ve her gebelikte %25 oranında etkilenmiş bir çocuk doğurma riski vardır. Genetik testler, taşıyıcılık durumu hakkında bilgi verebilir ve doğum öncesi testler yapılabilir.
Aileler İçin Öneriler
Çocuğunuzun gelişim sürecini düzenli olarak takip edin ve gerekli destekleri sağlayın.
Uyku ve davranış bozukluklarını yönetmek için bir uzmandan destek alın.
Çocuğun fiziksel sağlığını korumak için düzenli kontroller yaptırın.
Genetik danışmanlık alarak gelecekteki gebelikler hakkında bilgi edinin.
Destek gruplarına katılarak deneyimlerinizi paylaşın ve psikolojik destek alın.
Sonuç: MPS III, ilerleyici bir hastalık olup ailelerin bilinçli olması, erken müdahale ve destekleyici bakım ile çocuğun yaşam kalitesini artırabilir. Uzman doktorlar ve genetik danışmanlarla iş birliği içinde olmak, ailelerin en iyi kararları almasına yardımcı olacaktır.
Maroteaux-Lamy Sendromu, N-asetilgalaktozamin 4-sülfataz (Arylsulfataz B) enziminin eksikliği veya yokluğu nedeniyle ortaya çıkan nadir bir genetik hastalıktır.
Bu enzim eksikliği, glikozaminoglikanlar (GAG) olarak bilinen kompleks şeker zincirlerinin hücre içindeki lizozomlarda birikmesine yol açar. Sonuç olarak, çeşitli organ ve dokularda ilerleyici hasarlar meydana gelir.
Belirtiler: Hastalığın belirtileri, GAG birikiminin miktarına ve biriktiği dokulara bağlı olarak değişkenlik gösterir. Semptomlar genellikle çocukluk döneminde başlar ve zamanla ilerler. MPS VI’nın tipik belirtileri şunlardır:
İskelet Sistemi:
Boy kısalığı; büyüme genellikle 10 yaşına kadar durur.
Çoğu MPS hastalığının aksine, MPS VI zeka gelişimini olumsuz etkilemez.
Kalıtım Özellikleri: MPS VI, otozomal resesif bir hastalıktır. Bu, hastalığın ortaya çıkması için bireyin her iki ebeveynden de kusurlu geni alması gerektiği anlamına gelir.
Her hamilelikte, çocuğun bu geni taşıyan ebeveynlerden alarak hastalıktan etkilenme ihtimali %25’tir.
Tanı Yöntemleri: MPS VI tanısı genellikle gecikir veya yanlış konabilir. Doğru tanı için aşağıdaki testler uygulanır:
İdrar Testi: İdrarda GAG miktarı ölçülür. MPS VI hastalarının idrarında anormal derecede yüksek GAG seviyeleri tespit edilir.
Enzim Aktivite Testi: Kan örneği veya deri biyopsisinden elde edilen hücrelerde N-asetilgalaktozamin 4-sülfataz enziminin aktivitesi ölçülür. MPS VI hastalarında bu enzimin aktivitesi düşüktür.
Genetik Testler: ARSB genindeki mutasyonları tespit etmek için genetik analiz yapılabilir.
Doğum öncesi tanı için aile öyküsü olan bireylerde, anne karnındaki bebeğin enzim aktivitesi veya genetik yapısı incelenerek tarama yapılabilir.
Tedavi Seçenekleri: MPS VI’nın kesin bir tedavisi olmamakla birlikte, hastalığın belirtilerini hafifletmek ve ilerlemesini yavaşlatmak için çeşitli yöntemler mevcuttur:
Enzim Replasman Tedavisi (ERT): Galsülfaz (Naglazyme®) adlı enzim replasman tedavisi, 2005 yılında FDA tarafından onaylanmıştır. Bu tedavi, eksik olan enzimin yerine konmasını sağlar ve hastalığın ilerlemesini yavaşlatabilir.
Destekleyici Tedaviler:
Fizik Tedavi: Eklem hareketliliğini artırmak ve kas güçsüzlüğünü azaltmak için uygulanır.
Cerrahi Müdahaleler: İskelet anormalliklerini düzeltmek, solunum yollarını açmak veya görme problemlerini gidermek için çeşitli cerrahi işlemler yapılabilir.
Solunum Desteği: Solunum güçlüğü yaşayan hastalar için destekleyici cihazlar kullanılabilir.
Erken tanı ve tedavi, hastalığın seyrini olumlu yönde etkileyebilir ve yaşam kalitesini artırabilir.
Dikkat Edilmesi Gereken Hususlar:
Enzim Replasman Tedavisi (ERT): Mümkünse, tanı konulduktan hemen sonra ERT’ye başlanmalıdır. ERT, hastalığın ilerlemesini yavaşlatmada önemli rol oynar.
Düzenli Multidisipliner İzlem: Hastalığın multisistemik etkileri nedeniyle, farklı uzmanlık alanlarının bir araya geldiği bir sağlık ekibi tarafından düzenli olarak izlenmelidir.
Kas-İskelet Sistemi İzlemi: Eklem sertliği, büyüme anomalileri ve iskelet deformiteleri gibi bulguların erken tespiti ve yönetimi için ortopedi uzmanlarıyla düzenli kontroller yapılmalıdır.
Solunum Fonksiyonları: Üst ve alt solunum yolu obstrüksiyonları sık görüldüğünden, pulmonoloji uzmanları tarafından solunum fonksiyon testleri ve gerekli durumlarda destekleyici tedaviler planlanmalıdır.
Kardiyovasküler İzlem: Kalp kapak hastalıkları ve kardiyomiyopati riski nedeniyle, kardiyoloji uzmanları tarafından düzenli ekokardiyografi ve diğer kardiyak değerlendirmeler yapılmalıdır.
Göz Sağlığı: Korneal bulanıklık ve glokom gibi göz problemlerinin erken tespiti için oftalmoloji kontrolleri önemlidir.
İşitme Değerlendirmesi: İşitme kaybı riski nedeniyle, odyolojik testler ve KBB uzmanı değerlendirmeleri düzenli olarak yapılmalıdır.
Diş Sağlığı: Diş ve çene anomalileri sık görüldüğünden, düzenli diş hekimi kontrolleri ve gerekli ortodontik müdahaleler planlanmalıdır.
Anestezi ve Cerrahi İşlemler: Anestezi ve cerrahi işlemler, MPS VI hastalarında yüksek risk taşıdığından, deneyimli ekipler tarafından ve gerekli önlemler alınarak gerçekleştirilmelidir.
Takip Edilmesi Gereken Tıbbi Bölümler:
Genetik ve Metabolizma Uzmanları: Hastalığın tanısı, genetik danışmanlık ve tedavi planlaması için.
Ortopedi: İskelet ve eklem sorunlarının yönetimi için.
Pulmonoloji: Solunum yolu problemlerinin izlenmesi ve tedavisi için.
Kardiyoloji: Kalp ile ilgili komplikasyonların takibi ve yönetimi için.
Oftalmoloji: Göz problemlerinin erken tespiti ve tedavisi için.
Kulak Burun Boğaz (KBB): İşitme ve üst solunum yolu sorunlarının yönetimi için.
Diş Hekimliği ve Ortodonti: Ağız ve diş sağlığının korunması ve çene anomalilerinin düzeltilmesi için.
Anesteziyoloji: Cerrahi işlemler öncesi ve sırasında güvenli anestezi uygulamaları için.
Bu multidisipliner yaklaşım, MPS VI hastalarının yaşam kalitesini artırmak ve hastalığın ilerlemesini yavaşlatmak için kritik öneme sahiptir.
Maroteaux-Lamy Sendromu (Mukopolisakkaridoz Tip VI, MPS VI) tanı ve takibi için merkezimize başvurabilirsiniz. Merkez bilgilerimiz için tıklayınız.
Mukopolisakkaridoz Tip 4 (MPS Tip 4), nadir görülen genetik bir hastalıktır ve Morquio Sendromu olarak da bilinir. Bu hastalık, vücudun bazı karbonhidratları (glikozaminoglikanları – GAG’ları) parçalayamaması sonucu ortaya çıkar. Bu durum, özellikle iskelet sistemi ve bağ dokularında birikime neden olarak ilerleyici kemik bozukluklarına yol açar.
MPS Tip 4’ün Nedenleri
MPS Tip 4, vücuttaki belirli enzimlerin eksikliğinden kaynaklanır.
MPS 4B: GLB1 (β-galaktosidaz) enziminin eksikliğinden kaynaklanır. Bu enzimlerin eksik olması, keratan sülfat (KS) ve kondroitin-6-sülfat (C6S) adı verilen maddelerin vücutta birikmesine neden olur.
MPS Tip 4, otozomal resesif olarak kalıtılır, yani anne ve babadan her ikisi de taşıyıcı olduğunda çocuk hasta olabilir.
MPS Tip 4’ün Belirtileri
MPS Tip 4 belirtileri genellikle çocukluk çağında ortaya çıkar ve zamanla ilerleyebilir. 1. Kemik ve İskelet Sistemi Problemleri: ✔ Boy kısalığı (özellikle gövde kısa kalır) ✔ Omurgada eğrilikler (kifoskolyoz) ✔ Boyun omurlarında instabilite (C1-C2 düzeyinde kayma riski) ✔ Göğüs kafesinde şekil bozuklukları (pektus karinatum) ✔ Eklemlerde aşırı esneklik ve instabilite ✔ Dizlerde eğrilik (genu valgum)
2. Solunum ve Kardiyak Problemler: ✔ Hava yollarında daralma, solunum güçlüğü ✔ Uyku apnesi (gece solunum durmaları) ✔ Kalp kapak hastalıkları (mitral ve aort kapak yetersizliği)
3. Göz, Kulak ve Diş Problemleri: ✔ Kornea bulanıklığı (görme bulanıklığı yapabilir) ✔ İşitme kaybı (orta ve iç kulak etkilenebilir) ✔ Dişlerde şekil bozuklukları ve erken diş kayıpları
💡 Zeka Gelişimi: MPS Tip 4 hastalarında zeka genellikle normaldir, ancak omurilik veya sinir basıları nedeniyle ilerleyen yaşlarda hareket kısıtlılıkları görülebilir.
MPS Tip 4 Tanısı Nasıl Konur?
✔ İdrar testi: Keratan sülfat seviyeleri ölçülür. ✔ Enzim testi: GALNS veya GLB1 enzim aktiviteleri kontrol edilir. ✔ Genetik test: Kesin tanı için DNA analizi yapılır. ✔ Röntgen ve MR: Kemik anomalileri ve servikal instabilite değerlendirilir.
MPS Tip 4 Tedavi Yöntemleri
MPS Tip 4 için kesin bir kalıcı tedavi henüz yoktur, ancak bazı tedaviler hastaların yaşam kalitesini artırabilir.
1️⃣ Enzim Replasman Tedavisi (ERT):
Elosulfaz alfa (Vimizim®), MPS 4A için FDA onaylıdır.
Kemik bozukluklarını tamamen düzeltemese de hastalığın ilerleyişini yavaşlatabilir.
Erken dönemde başlanması önerilir.
2️⃣ Destekleyici Tedaviler:
Fizik tedavi ve rehabilitasyon: Eklem hareketlerini koruyarak kas gücünü artırmak için gereklidir.
Ortopedik ameliyatlar: Diz, omurga veya boyun instabilitesini düzeltmek için gerekebilir.
Solunum desteği: CPAP/BiPAP cihazları gece solunum sıkıntısı olan hastalara yardımcı olabilir.
Kardiyak takip: Kalp kapak hastalıkları için düzenli kontroller gereklidir.
MPS Tip 4’ün Seyri ve Yaşam Beklentisi
Hastalığın şiddeti kişiden kişiye değişebilir. ✔ Hafif olgularda, bireyler yetişkinliğe kadar bağımsız hareket edebilir. ✔ Şiddetli olgularda, genellikle 20-30’lu yaşlarda tekerlekli sandalyeye bağımlı hale gelinir. ✔ Ana ölüm nedenleri solunum yetmezliği ve kardiyak komplikasyonlardır.
Aileler İçin Öneriler
✔ Multidisipliner takip önemlidir. Çocuk metabolizma uzmanı, ortopedi, kardiyoloji ve solunum hastalıkları uzmanları süreci birlikte yönetmelidir. ✔ Fiziksel aktiviteler desteklenmelidir ancak eklem stabilitesine dikkat edilmelidir. ✔ Uyku apnesi ve solunum problemleri için erken önlem alınmalıdır.
Mukopolisakkaridoz Tip 2 (MPS Tip 2), lizozomal depo hastalıkları grubuna ait, nadir görülen bir genetik hastalıktır. Hunter Sendromu olarak da bilinir ve X’e bağlı resesif geçiş gösterir. Bu nedenle genellikle erkeklerde görülür ancak nadir de olsa kız vakalar bildirilmiştir. Hastalık, iduronat-2-sülfataz (I2S) enziminin eksikliği nedeniyle glikozaminoglikanların (GAG) hücrelerde birikmesi ile ortaya çıkar.
Belirtileri Nelerdir?
MPS Tip 2 belirtileri, hastalığın şiddetine göre değişebilir. Hafif ve ağır olmak üzere iki alt tipi bulunur:
Hafif Form: Zeka genellikle normaldir, ancak eklem sertliği, kaba yüz hatları, işitme kaybı, dalak ve karaciğer büyüklüğü gibi semptomlar görülür.
Ağır Form: Zihinsel gelişim geriliği, belirgin yüz anormallikleri, eklem sertliği, büyüme geriliği ve solunum sorunları gibi ciddi belirtiler içerir.
Genel belirtiler:
Kaba yüz görünümü (geniş alın, burun kökü basıklığı, kalın dudaklar)
Eklem sertliği ve hareket kısıtlılığı
Karaciğer ve dalak büyüklüğü (hepatosplenomegali)
Solunum problemleri ve sık tekrarlayan enfeksiyonlar
Kardiyovasküler sorunlar
Davranış bozuklukları ve nörolojik etkilenme (ağır formda)
İşitme kaybı
Hidrosefali
Papüler cilt lezyonları, sırtta mongol lekeleri
Gastrointestinal sistem etkilenmesi (ishal vb.)
MPS Tip 2 Nasıl Teşhis Edilir?
MPS Tip 2 tanısı, klinik bulgulara dayanarak ve aşağıdaki testlerle doğrulanır:
İdrarda glikozaminoglikan (GAG) testi (Dermatan sülfat ve heparan sülfat birikimi)
Enzim aktivite testi (İdrar, kan veya fibroblastlarda iduronat-2-sülfataz enzim düzeylerinin ölçülmesi)
Genetik analiz (IDS gen mutasyonu tespiti)
Tedavi Yöntemleri Nelerdir?
MPS Tip 2 tedavisinde çeşitli yöntemler mevcuttur:
Enzim Replasman Tedavisi (ERT): Rekombinant iduronat-2-sülfataz (Idursulfase – Elaprase®) ile tedavi edilir. Haftalık 0.5 mg/kg/doz infüzyon uygulanır. ERT, yürüme mesafesini artırabilir, dalak ve karaciğer büyüklüğünü azaltabilir ancak nörolojik etkilenme üzerinde sınırlı etkisi vardır. Ayrıca Hunterase® (Idursulfase beta) adlı ilaç da benzer şekilde kullanılmaktadır.
Hematopoetik Kök Hücre Nakli (HSCT): Sonuçları değişken olmakla beraber, gelişimi koruma açısından faydalı olabilir. Ancak ortopedik ve göz bulgularında belirgin iyileşme sağlanamamıştır. Transplantasyona bağlı komplikasyon oranı yüksektir.
Semptomatik Tedaviler:
Fizyoterapi ve eklem hareket açıklığını koruyucu tedaviler
Solunum desteği ve enfeksiyon kontrolü
Kardiyovasküler takip ve gerekli durumlarda cerrahi girişimler
İşitme kaybı için işitme cihazları veya koklear implantlar
Gastrointestinal destek ve beslenme tedavileri
İntratekal Uygulamalar: Beyin-bariyerini aşamayan ERT’nin etkisini artırmak amacıyla intratekal uygulamalara yönelik araştırmalar devam etmektedir.
Hasta Yakınları İçin Öneriler
Düzenli Takip: Multidisipliner bir ekip tarafından takip edilmesi gereklidir. Aşağıdaki takip çizelgesi uygulanabilir:
Alfa Mannosidoz, alfa-mannosidaz enziminin eksikliği ile karakterize nadir görülen bir genetik hastalıktır. Bu durum kompleks şeker moleküllerinin çeşitli organ ve dokularda birikmesine yol açarak çok çeşitli semptomlara yol açar.
Alfa Mannosidoz, MAN2B1 genindeki mutasyonların neden olduğu otozomal resesif bir hastalıktır. Bir çocuğun etkilenmesi için her iki ebeveyn de mutasyona uğramış genin bir kopyasını taşımalıdır.
Klinik Özellikler
Alfa Mannosidoz, gelişimsel gecikmeler, zihinsel yetersizlik, iskelet anormallikleri, işitme kaybı, tekrarlayan enfeksiyonlar, yüzde kabalaşma ve organomegali (genişlemiş organlar) dahil olmak üzere bir dizi semptomla kendini gösterir.
1.Zihinsel Yetersizlik ve Gelişimsel Gecikmeler:
Alfa Mannosidozlu çocuklar genellikle çeşitli derecelerde zihinsel yetersizlik yaşarlar. Gecikmiş konuşma ve motor beceriler de dahil olmak üzere gelişimsel dönüm noktalarını geciktirmiş olabilirler.
2.Yüz Kabalaşması ve İskelet Anormallikleri:
Zamanla, Alfa Mannosidozlu bireyler belirgin bir alın, kaba yüz özellikleri, geniş bir burun ve büyük bir dil gibi belirgin yüz özellikleri geliştirebilir. Genişlemiş eklemler, kalınlaşmış kemikler ve boy kısalığı gibi iskelet anormallikleri de mevcut olabilir.
3.İşitme kaybı:
İşitme bozukluğu, Alfa Mannosidozun ortak bir özelliğidir. Hafif ila şiddetli arasında değişebilir ve gecikmiş konuşma gelişimine neden olabilir.
4.Tekrarlayan Enfeksiyonlar:
Bozulmuş bağışıklık fonksiyonu nedeniyle, Alfa Mannosidozlu bireyler, özellikle solunum ve idrar yollarında sık sık enfeksiyonlar yaşayabilir. Bu tekrarlayan enfeksiyonlar, doğada bakteriyel, viral veya mantar olabilir.
5.Organomegali:
Organomegali olarak bilinen genişlemiş organlar, Alfa Mannosidozda ortaya çıkabilir. Karaciğer ve dalak en sık etkilenir ve hepatomegali (genişlemiş karaciğer) ve splenomegali (genişlemiş dalak) ile sonuçlanır.
6.Nörolojik Belirtiler:
Alfa Mannosidoz ilerledikçe, nörolojik semptomlar daha belirgin hale gelebilir. Bunlar arasında ataksi (bozulmuş koordinasyon), kas zayıflığı, titreme, nöbetler ve denge ve yürüme ile ilgili sorunlar yer alabilir.
7.Görüş problemleri:
Alfa Mannosidozlu bazı bireyler, görme keskinliğini etkileyebilecek korneanın bulanıklaşması (kornea opaklığı) veya retina dejenerasyonu gibi görme sorunları geliştirebilir.
Teşhis ve Hastalık Yönetimi:
Alfa Mannosidozun teşhisi klinik değerlendirme, enzim aktivite testi ve genetik testi içerir. Erken teşhis, zamanında müdahale ve destek için çok önemlidir.
Hematopoietik kök hücre nakli (HSCT), alfa-mannosidaz enzim eksikliğinden kaynaklanan nadir bir genetik bozukluk olan Alfa Mannosidozu olan kişiler için potansiyel bir tedavi seçeneğidir. Kemik iliği nakli olarak da bilinen HSCT, kusurlu veya eksik hücrelerin yerine sağlıklı kök hücrelerin hastaya verilmesini içerir.
HSCT, eksik enzimi üreten bir hücre kaynağı sağlayarak Alfa Mannosidozun altında yatan nedeni kontrol altına alma potansiyeli sunar. Uyumlu bir vericiden (allojenik transplantasyon) veya hastanın kendisinden (otolog transplantasyon) elde edilebilen nakledilen kök hücreler, kemik iliğine göç etme ve alfa üretmekten sorumlu olanlar da dahil olmak üzere çeşitli kan hücresi tiplerine farklılaşma kapasitesine sahiptir.
Bireysel bir hasta için HSCT’nin uygunluğunu ve potansiyel faydalarını belirlemek için HSCT’de ve Alfa Mannosidozun yönetiminde deneyimli uzman bir tıbbi ekibe danışmak çok önemlidir. Ayrıntılı bilgi sağlayacak, riskleri ve olası sonuçları tartışacak ve karar verme sürecine rehberlik edeceklerdir.
HSCT’nin Alfa Mannosidozlu bireylerde mevcut nörolojik hasarı tam olarak tersine çeviremeyebileceğini ve etkinliğinin kişiden kişiye değişebileceğini akılda tutmak önemlidir.
Velmanase alfa, biyoteknoloji ile üretilen bir enzimdir. Velmanase alfa, eksik veya kusurlu alfa-mannosidaz enzimini yerine geçerek çalışır. Bu ilacı intravenöz olarak uygulayarak vücuda eksik olan enzimi sağlar ve birikmiş kompleks şekerlerin parçalanmasını kolaylaştırır.
Velmanase alfa ile tedavi, semptomları hafifletmeyi ve Alpha Mannosidozun ilerlemesini yavaşlatmayı amaçlar. İskelet anormallikleri, organomegali (büyümüş organlar) ve genel yaşam kalitesi gibi durumun belirli yönlerini iyileştirmeye veya stabilize etmeye yardımcı olabilir. Bununla birlikte, velmanase alfa’nın mevcut nörolojik hasarı tersine çevirmeyebileceğini veya Alpha Mannosidoz ile ilişkili tüm semptomları tamamen ortadan kaldıramayacağını not etmek önemlidir.
Tıbbi Bakım: Düzenli tıbbi takipler, durumun ilerlemesini izlemek için çok önemlidir. Tedavi, semptom yönetimini, kemik iliği nakli veya enzim replasman tedavisini ve spesifik komplikasyonları ele almak için destekleyici bakımı içerebilir.
Eğitim Desteği ve Gelişimsel Müdahaleler:
Erken Müdahale: Konuşma terapisi, mesleki terapi ve fizik tedaviyi içeren erken müdahale programları, çocuğun genel gelişimini destekleyebilir ve fonksiyonel yeteneklerini geliştirebilir.
Özel Eğitim: Bireyselleştirilmiş bir eğitim planı geliştirmek için özel eğitim uzmanlarıyla işbirliği yapmak, akademik ve sosyal ihtiyaçlar için özel olarak hazırlanmış destek sağlayabilir.
Sağlıklı Yaşam Tarzı: Besleyici diyet seçenekleri, düzenli fiziksel aktivite (uygun olduğu şekilde) ile sağlıklı bir yaşam tarzını teşvik etmek, genel refah için çok önemlidir.
Çevresel Değişiklikler: Güvenli ve erişilebilir bir ev ortamı oluşturmak, potansiyel tehlikeleri en aza indirmek ve Alfa Mannosidozlu bireyler için optimum hareketliliği sağlamak için önemlidir.











































Yorum yazabilmek için oturum açmalısınız.