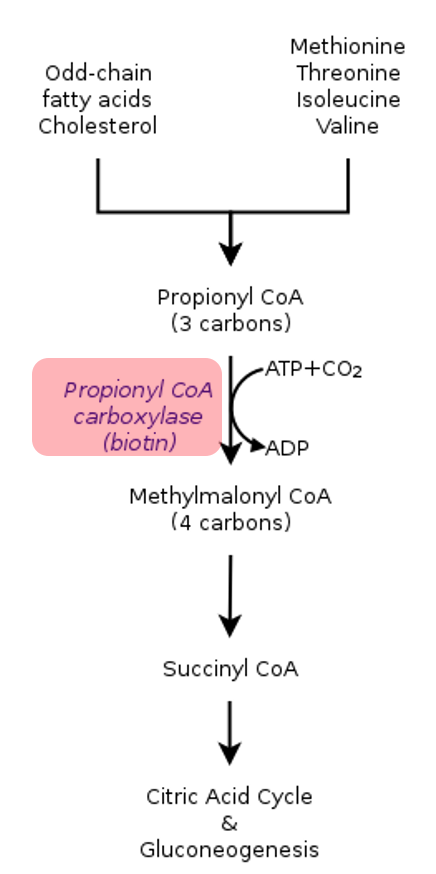
Besinlerle alınan ‘’Valin, metionin, treonin ve izolösin’’ adı verilen amino asitlerin ve yağ asitlerinin metabolize olmaması sonucu ortaya çıkan bir organik asidemidir.
Anne ve babadan genetik (otozomal resesif) olarak geçer.
Hastalar ‘’Valin, metionin, treonin ve izolösin’’ adı verilen amino asitlerin ve yağ asitlerini ihtiva eden besin ile beslendikten sonra metabolitleri olan propiyonik asit ve glisin kanda birikerek hastalık bulgularına yol açar.
Hastalık Propionil CoA karboksilaz adı verilen bir enzimin yeterli işlevi görmemesinden kaynaklanır.
KLİNİK
Genellikle bulgular yenidoğan döneminde başlamakla birlikte hastalığın hafif formlarında ilk bulgular çocukluk veya adölesan dönemde de görülebilir.
Hastalar kusma, beslenememe, halsizlik, uyku hali ile gelebilir.
Ağır hastalarda nöbet ve koma ve inme görülebilir.
Metabolik krizer yüksek proteinli gıda ile beslenme, uzun süre açlık, ateşli enfeksiyonlar ve cerrahi operasyonlar ile tetiklenebilir.
Metabolik kriz dönemleri arasında propiyonik asidemili çocuklar sağlıklı olabilir. Ancak, bazılarının nörolojik gelişme geriliği ve farklı oraganları ilgilendiren sağlık sorunları görülebilir. Bazı hastaların, metabolik kriz yaşamamış olsalar bile uzun vadeli sorunları vardır. Bunlar:
Öğrenme sorunları veya zihinsel engeller
Yürüme ve motor becerilerdeki gecikmeler
Anormal istemsiz hareketler (distoni ve kore)
Spastisite adı verilen sert kas tonusu
Kısa boylu zayıf büyüme
Deri döküntüleri ve enfeksiyonları
Osteoporoz
Karaciğer büyümesi
Kalp problemleri
Böbrek hastalığı veya yetmezliği
Gözdeki sinirlerle ilgili problemlere bağlı görme kaybı
LABORATUVAR
Hastaların eş zamanlı laboratuvar bulgularında metabolik asidoz, kan şekeri düşüklüğü, kanda ve idrarda keton artışı, kanda amonyak yüksekliği, anemi, kan pulcuklarında (trombosit) ve akyuvar sayısında azalma görülebilir.
Ataklar arasında ve hastalığın hafif olduğu hastalarda semptom ve bulgular görülmez.
TANI
Tandem MS ve idrar organik asitlerindeki metabolik atılımlar ile tanı konulabilir.
Genetik analiz (PCCA, PCCB) ile de tanı konulabilmektedir.
TEDAVİ
Zihinsel engellilik olasılığını ve ciddi tıbbi problemleri azaltmak için erken tedavi elzemdir.
Ağızdan alınan antibiyotikler, bağırsak bakterileri tarafından yapılan propiyonik asit miktarını azaltmaya yardımcı olabilir. Hastanın antibiyotiğe ihtiyacı olup olmadığına klinik takibe göre karar verilir.
Bazı hastalara ağız yoluyla biotin takviyesi verilebilir. Biotin, vücudun gıdalardan enerji elde etmesine yardımcı olan bir B vitamini türüdür. Biotin’in Propionil CoA karboksilaz enzimine yardımcı olduğu kanıtlanmamıştır, ancak hastalar için yararlı olup olmadığını görmek için denebilen bir tedavidir.
İzolösin, valin, metiyonin ve treonin amino asitlerinde düşük miktarda içeren diyet tedavisi başlanmalıdır. Diyetteki çoğu yiyecek karbonhidrat ağırlıklıdır (ekmek, tahıl, makarna, meyve, sebze vb.). Karbonhidrat oranı yüksek, protein ve yağ oranı düşük bir diyet, metabolik krizlerin önlenmesine yardımcı olabilir.
L-kartinitin hastalarda propiyonik asidin uzaklaştırılması için verilebilmektedir.
Akut krizlerde damar içi yüksek glikoz ve bikarbonat tedavisi gerekebilir.
Amonyak düşürücü tedavilere yanıt alınamaması halinde diyaliz uygulanabilir.
Besinlerle alınan ‘’Valin, metionin, treonin ve izolösin’’ adı verilen amino asitlerin ve yağ asitlerinin metabolize olmaması sonucu ortaya çıkan bir organik asidemidir.
Anne ve babadan genetik (otozomal resesif) olarak geçer.
Hastalar ‘’Valin, metionin, treonin ve izolösin’’ adı verilen amino asitlerin ve yağ asitlerini ihtiva eden besin ile beslendikten sonra metabolitleri olan metilmalonik asit ve glisin kanda birikerek hastalık bulgularına yol açar.
Birkaç farklı MMA türü vardır. Bazı türler B12 vitamini enjeksiyonları ile tedavi edilebilir. Bu türlere “B12 vitamini duyarlı” denir. Genellikle B12 vitamini ile tedavi edilebilen iki MMA türü, Kobalamin A (CblA) eksikliği ve Kobalamin B (CblB) eksikliğidir.
B12 vitamini ile tedavi edilemeyen başka MMA türleri de vardır. Bu türlere “yanıt vermeyen B12 vitamini” adı verilir. Bunlardan biri “Mut 0” olarak adlandırılır. Metilmalonil-CoA mutazadı verilen bir enzimin bulunmamasından kaynaklanır. B12 vitamini tedavisine yanıt vermeyen başka bir MMA türü, “Mut-” olarak adlandırılır. “Mut-” tipi MMA’ya sahip kişilerde çok az metilmalonil-CoA mutaz enzimi vardır.
KLİNİK
Genellikle bulgular yenidoğan döneminde başlamakla birlikte hastalığın hafif formlarında ilk bulgular çocukluk veya adölesan dönemde de görülebilir.
Hastalar kusma, beslenememe, halsizlik, uyku hali ile gelebilir.
Ağır hastalarda nöbet ve koma ve inme görülebilir.
Metabolik krizer yüksek proteinli gıda ile beslenme, uzun süre açlık, ateşli enfeksiyonlar ve cerrahi operasyonlar ile tetiklenebilir.
Metabolik kriz dönemleri arasında MMA’lı çocuklar sağlıklı olabilir. Ancak, bazılarının nörolojik gelişme geriliği ve farklı organları ilgilendiren sağlık sorunları görülebilir. Bazı hastaların, metabolik kriz yaşamamış olsalar bile uzun vadeli sorunları vardır. Bunlar:
Öğrenme sorunları veya zihinsel engeller
Yürüme ve motor becerilerdeki gecikmeler
Anormal istemsiz hareketler (distoni ve kore)
Spastisite adı verilen sert kas tonusu
Kısa boylu zayıf büyüme
Deri döküntüleri ve enfeksiyonları
Osteoporoz
Karaciğer büyümesi
Kalp problemleri
Böbrek hastalığı veya yetmezliği
Gözdeki sinirlerle ilgili problemlere bağlı görme kaybı
MMA’lı az sayıda hastada hiçbir zaman semptom görülmez.
LABORATUVAR
Hastaların eş zamanlı laboratuvar bulgularında metabolik asidoz, kan şekeri düşüklüğü, kanda ve idrarda keton artışı, kanda amonyak yüksekliği, anemi, kan pulcuklarında (trombosit) ve akyuvar sayısında azalma görülebilir.
Ataklar arasında ve hastalığın hafif olduğu hastalarda semptom ve bulgular görülmez.
TANI
Tandem MS ve idrar organik asitlerindeki metabolik atılımlar ile tanı konulabilir.
Genetik analiz (MUT, MMAA, MMAB) ile de tanı konulabilmektedir.
TEDAVİ
Zihinsel engellilik olasılığını ve ciddi tıbbi problemleri azaltmak için erken tedavi elzemdir. B12 vitaminine duyarlı MMA olan çocuklara B12 vitamini verilir. Ek olarak, çoğu çocuğun düşük proteinli bir diyete girmesi ve özel bir tıbbi formül başlanması gerekir. Hastada MMA olduğunu öğrenir öğrenmez tedavilere başlamalıdır.
CblA eksikliği olan çocukların % 90’ından fazlası B12 vitamini tedavisine yanıt verir. CblB eksikliği olan çocukların yaklaşık % 40’ına bu tedavi yardımcı olabilir.
Ağızdan alınan antibiyotikler, bağırsak bakterileri tarafından yapılan metilmalonik asit miktarını azaltmaya yardımcı olabilir. Hastanın antibiyotiğe ihtiyacı olup olmadığına klinik takibe göre karar verilir.
İzolösin, valin, metiyonin ve treonin amino asitlerinde düşük miktarda içeren diyet tedavisi başlanmalıdır. Diyetteki çoğu yiyecek karbonhidrat ağırlıklıdır (ekmek, tahıl, makarna, meyve, sebze vb.). Karbonhidrat oranı yüksek, protein ve yağ oranı düşük bir diyet, metabolik krizlerin önlenmesine yardımcı olabilir.
L-kartinitin hastalarda metilmalonik asidin uzaklaştırılması için verilebilmektedir.
Akut krizlerde damar içi yüksek glikoz ve bikarbonat tedavisi gerekebilir.
Amonyak düşürücü tedavilere yanıt alınamaması halinde diyaliz uygulanabilir.
Besinlerle alınan ‘’lösin’’ adı verilen amino asidin metabolize olmaması sonucu ortaya çıkan bir organik asidemidir.
Anne ve babadan genetik (otozomal resesif) olarak geçer (IVD geni).
Bir çok metabolik hastalıkta olduğu gibi izovalerik asidemide de taşıyıcı anne ve taşıyıcı babanın sorunlu allellerinin çocuklarında yan yana gelmesi ile oluşur. Taşıyıcı anne babanın çocuklarının hasta olma ihtimali %25 iken %50 ihtimalle çocukları taşıyıcı olur ve %25 sağlam çocuk sahibi olma ihtimalleri vardır. Akraba evlilikleri bu sorunlu allellerin yan yana gelme ihtimalini arttırmaktadır.
Hastalarda ‘’Lösin’’i metabolize eden ‘’izoveleril KoA dehidrogenaz’’ enziminin sentezinde sorun vardır. Hastalar lösin ihtiva eden besin ile beslendikten sonra lösin metabolitleri kanda birikerek hastalık bulgularına yol açar.
KLİNİK
Genellikle bulgular yenidoğan döneminde başlamakla birlikte hastalığın hafif formlarında ilk bulgular çocukluk veya adölesan dönemde de görülebilir.
Hastalar kusma, beslenememe, halsizlik, uyku hali ile gelebilir.
Vücutta terli ayak kokusu önemli bulgulardan biridir.
Ağır hastalarda nöbet ve koma görülebilir.
Hastaların eş zamanlı laboratuvar bulgularında metabolik asidoz, kan şekeri düşüklüğü, kanda ve idrarda keton artışı, kanda amonyak yüksekliği, anemi, kan pulcuklarında (trombosit) ve akyuvar sayısında azalma görülebilir.
Ataklar arasında ve hastalığın hafif olduğu hastalarda semptom ve bulgular görülmez.
TANI
Tandem MS ve idrar organik asitlerindeki metabolik atılımlar ile tanı konulabilir.
Genetik analiz (IVD geni) ile de tanı konulabilmektedir.
TEDAVİ
Tedavide düşük proteinli lösin içeriği düşük diyet önerilmektedir. Bu diyet için özel lösin içermeyen enteral ürünler verilmektedir.
L-glisin tedavisi vücuttaki izovalerik asitlerin uzaklaştırılması için verilmektedir.
L-karnitin de bazı hastalarda izovalerik asidin uzaklaştırılması için verilebilmektedir.
Akut krizlerde damar içi yüksek glikoz ve bikarbonat tedavisi gerekebilir.
Amonyak düşürücü tedavilere yanıt alınamaması halinde diyaliz uygulanabilir.
UZUN DÖNEM İZLEM
Erken tanı ve tedavi alan akut krizlere girmesi önlenen hastalarda normal büyüme ve gelişme ile sağlıklı bir yaşam sürdürebilmektedir.
Çocuklar büyüdükçe daha az metabolik kriz atakları geçirme eğilimindedirler.
Ağır ve tekrarlayan ataklar yaşam boyu öğrenme sorunlarına veya zihinsel engelliğe yol açabilir.
Fenilketonüri tanılı annelerin gebelikleri sırasında, kandaki yüksek fenilalanin seviyesinin anne karnındaki bebeği etkilemesi durumuna maternal fenilketonüri denilmektedir.
Bebekte doğum kilosunda düşüklük, baş çevresi küçüklüğü, anormal yüz bulgular, büyüme gelişme geriliği, göz ve kalp sorunları ve zeka geriliği görülebilir.
Hafif hiperfenilalaninemili annelerde bu risk daha düşüktür. Fakat hiperfenilalaninemi tanılı annelerin de gebelikleri süresince mutlaka kan fenilalanin düzeyi ile takibi gerekmektedir.
Bu durumun oluşmaması için gebelik öncesi (en az 3 ay öncesinden) kan fenilalanin düzeyi 2-6 mg/dl (120-360mmol/L) arasında tutulmalıdır. Bu olgularda diyet tedavisinden dolayı meydana gelen vitamin ve mineral desteği sağlanmalıdır.
Gebelik boyunca kan fenilalanin düzeyi 2-4 mg/dl (120-240mmol/L) arasında olacak şekilde sıkı takip edilmelidir.
Sıkı diyetin oluşturabileceği vitamin mineral eksiklikleri giderilmelidir.
Fabry Hastalığı X kromozumu üzerinden geçiş gösteren lizozomal depo hastalığıdır.
Hastalık dünyada 40000-117000 kişide 1 görülmektedir.
a-galaktosidaz A adlı enzim eksikliğine bağlı glikolipit birikimi [globotriaosilseramid (GL3)], farklı dokularda hücre içinde birikir.
Enzim eksikliğinin derecesine göre klinik bulgularda değişiklik gösterebilmektedir.
Klasik Fabry hastalığında; erken yaşlarda başlayan genellikle karın ve kasık bölgesindeki ciltte kırmızı kabarık döküntüler (anjiokeratoma korporis diffusum), ellerde uyuşma ve ağrı (akroparestezi), az terleme (terleyememe) (hipohidrozis), ve bunlara bağlı egzersiz ve sıcağa tahammülsüzlük ve ateş yüksekliği görülebilir. Sindirim sistemini de tutan bu hastalık hastalarda karın ağrısı, ishal yapabilir. Enzimin metabolize edemediği maddeler gözün korneasında birikerek ‘’cornea verticillata’’ diye adlandırılan lekelenmelere yol açabilmektedir.
Hastalığın en önemli bulguları ise genellikle30’lu yaşlardan sonra başlayan böbrek ve kalp etkilenmesidir.
Bunun yanında hastalarda kulak çınlaması (tinnutus) ve tekrarlayan inme atakları da görülebilmektedir.
Hastalık doğal haline bırakılırsa (tedavisiz izlemde) erkeklerde yaşam süresini ortalama 20 yıl bayanlarda ise ortalama 10 yıl kadar azalttığı belirtilmektedir.
TANI:
Erkek hastalarda enzim (GL3) eksikliğinin gösterilmesi veya genetik analiz yeterliyken kadınlarda genetik analiz yapılması gerekmektedir.
Vücutta eksik olan enzimin yerine konması tek bu hastalığın en önemli tedavi yöntemidir.
Enzimin yerine konulması tedavisi ile birlikte hastalığın ilerlemesi önlendiği ve bulgularda gerileme görüldüğü belirtilmektedir. Bu yüzden, hastalara erken tanı konulması ve erken tedavi başlanması hastalığın ilerlemesinin yavaşlatılmasında oldukça önemlidir.
KLİNİK:
Bir çoğu klasik ağır klinik bulgularla seyretmektedir.
Klasik formda enzim aktivitesi yok denecek kadar azdır.
Daha hafif ve geç başlangıçlı tipleri olan sadece kalbi (kardiyak) veya böbreği (renal) tutan formları da mevcuttur.
Kardiyak Varyant:
60-70li yaşlarda kalp kasında büyüme (ventrikül hipertrofisi) ile kendini gösterir.
Hastalarda kalpte aritmi de yapabilir (kalp içi elektrik iletim bozukluğu).
Bu hastalarda genellikle ağır böbrek etkilenmesi görülmemektedir.
Daha çok mRNA’yı kısaltan missense mutasyonlar veya intronik lezyonlar bu duruma neden olmaktadır.
Renal Varyant:
Bu hastalarda böbrekler tutlulur. Böbrek yetmezliğine kadar ilerleyebilir. Hastalar zamanında ve uygun tedavi edilemezse diyaliz bağımlı hale gelebilir veya böbrek nakli yapılması gerekebilir.
TEDAVİ:
Son yıllarda çeşitli tedavi yöntemleri gündeme gelmekle birlikte (şaperon tedavisi, substrat redüksiyon tedavisi [migalastat]) en yaygın ve etkin kullanılan yöntem eksik olan enzimin yerine konması (enzim replasman tedavisi) tedavisidir.
Hastalar belli aralıklarla damar içine verilecek şekilde enzim tedavisi almaları gerekmektedir.
Tedavinin başarısını belirleyen en önemli faktörler tedavinin erken başlanması ve tedavinin aksatılmadan yapılmasıdır.
İki farklı enzim [Agalsidase alpha (Replagal, Shire Takeda), Agalsidase beta (Fabrazyme, Genzyme)] kullanılmaktadır.
Ülkemizde üretilmeyen bu enzimler sağlık sigortası tarafından karşılanmaktadır. Hastalar bu tedavi için bir ücret ödememektedirler.
Tedavi başarısını etkileyen en önemli faktörler klinik bulguların erken döneminde tedaviye başlanmış olması ve tedavinin aksatılmadan düzenli alınmasıdır.
İZLEM PLANI
Hastalar; metabolizma, nefroloji, kardiyoloji, nöroloji, göz, psikiyatri, kulak-burun-boğaz, gastroenteroloji, gibi bölümlerle ortak izlenmesi gerekmektedir.
Fabry tanısı alan bir kişinin tüm akraba ve ailesi Fabry hastalığı taranmasının yapılması, hastalığın erken yakalanıp tedavisine başlanılması açısında oldukça önemlidir.
Hücrelerde bulunan lizozomlar adı verilen yapılar içinde yer alan asit α-glukozidaz enzim eksikliği nedeni ile ortaya çıkan genetik geçişli bir hastalıktır.
Anne ve babadan genetik (otozomal resesif) olarak geçer (GAA geni).
Klinik Belirtiler:
Hastalık temelde iskelet kaslarını, kalp kasını ve düz kasları tutar.
Hastalığın ağırlığı, başlangıç yaşına, organ tutulum derecesine ve kas tutulumunun ağırlığına bağlıdır.
Enzim eksikliği ne kadar fazla ise hastalık o kadar erken bulgu verir ve ağır seyreder.
Hastalığın temel bulguları, kalp büyümesi, kalp kası etkilenmesi, vücut kaslarında güçsüzlüğe bağlı baş tutma, desteksiz oturma gibi gelişim gerilikleri, beslenme güçlüğü, solunum yetmezliği, solunum yolu enfeksiyonlarıdır.
Enzim eksikliği düzeyine bağlı olarak bulguların görülme yaşına göre infantil form (doğumdan itibaren klinik bulguların ortaya çıktığı tip) (klasik form) ve geç başlangıçlı form (non-klasik form) olarak ikiye ayrılmaktadır.
Klasik Pompe Hastalığı (İnfantil/Erken) :
Enzmi aktivitesinin çok düşük olduğu ya da olmadığı hasta grubudur.
Bulgular ilk 1 ayda ortaya çıkar. Kas zayıflığı, baş tutmada gecikme gibi motor gerilik ve büyüme geriliği görülür.
Tedavi başlanmayan olgularda 1 yaşına varmadan kalp veya solunum yetmezliğinden kaybedilir.
Olgularda kanda bakılan kreatin kinaz (CK) yüksekliği mevcuttur.
Ekokardiyografi (EKO) ile kalp büyümesi görülebilir.
Elektromiyografide (EMG) kas etkilenmesine ait bulgular saptanabilir.
Non-Klasik Pompe Hastalığı (Geç başlangıçlı):
Erişkin döneme kadar her hangi bir dönemde bulgu verir.
Yavaş ilerleyen kas güçsüzlüğü vardır. Olgularda solunum kaslarının tutulumuna bağlı solunum yetmezliği görülebilir.
Hastalarda koşma, performans sporlarında tırmanma gibi aktivitelerde yetersizdir.
Halsizlik ve ağrı olguların yarında bildirilmiştir.
Kas bulguları olan olgularda mutlak solunum yetersizliği açısından değerlendirme yapılmalıdır. Uyku anında oluşabilecek solunum yetmezliği için polisomnografi yapılmalıdır.
Hastalık yavaş seyirlidir. Tedavi almayan olgularda ilerleyen dönemlerde hastalar tekerlekli sandalyeye bağımlı veya solunum cihazına bağımlı kalabilir.
Ölümün en sık nedeni solunum yetmezliği olarak bildirilmektedir.
Tanı:
Tanıda kreatin kinaz (CK) yüksekliği önemli bir bulgudur. Bunun yanında kas yıkımına bağlı ALT, AST ve LDH yüksekliği de görülebilir.
Göğüs filminde kalp büyüklüğü saptanabilir.
EKO’da kalp büyüklüğü bulguları mevcuttur. Kalp yetmezliği bulguları saptanabilir.
Enzim düzeyi tayini (kas biyopsisinden, fibroblasttan ya da lökositten) bakılabilir. Deri biyopsisinden fibroblast enzim aktivitesi bakılması enzim düzeyini koymada en iyi yöntemdir.
Kas biyopsisi enzim düzeyini göstermede ve mikroskobik incelemede PAS boyası ile glikojen depolarının ve kas harabiyetinin gösterilmesinde önemlidir. Harabiyet olan yerlerde fibroblast ve yağ dokusu gözlenir.
Non-klasik formların %20’sinin biyopsisinde hem depolanma hem de kas harabiyeti kas tutulmunun heterojenitesi nedeni ile gösterilemeyebilir.
İdrar analizinde tetrasakkaridlerin (Glc4) gösterilmesi Pompe hastalığı için çok duyarlı olmakla birlikte özgüllüğü çok düşüktür. Başka glikojen depo hastalıklarında da idrarda tetrasakkarid atılımı artmaktadır. Fakat tedaviye yanıtın değerlendirilmesinde önemli bir yöntemdir.
Genetik olarak tanı koymak da mümkündür.
Tedavi:
Tedavinin temelinde olası kalp ve solunumsal yetmezliklerin destek tedavisi yatmaktadır.
Alaninden zengin, yüksek proteinli diyet, tedavilerin kısmi faydalı olabildiği bildirilmektedir.
Kalp naklinin etkili olmayacağı öne sürülmektedir.
1999’da enzim replasman tedavisi geliştirilmesi hastalığın ilerleyişini önemli ölçüde yavaşlatmaktadır.
Enzim Replasman Tedavisi:
Myozyme® (alglukosidaz alfa) kullanılmaktadır. Ülkemize Türk Eczacılar Birliği üzerinden yurtdışından gelmektedir.
En dikkat edilmesi gereken konu tedavinin en erken şekilde başlanmasıdır.
Hastalık bulguları ağır olsa dahi enzim tedavisine erken başlandığında bulguların geri döndüğü görülmüştür.
Tedavi damar içine klinik durumuna göre 1-2 haftada bir verilmelidir.
Asit-b-glukosidaz enzim eksikliğine bağlı ortya çıkan hastalıktır. Glukoseramid adlı madde bu enzim ile glukoz ve seramide parçalanmaktadır. Enzim eksikliğinde ise vücudumuzun çeşitli yerlerindeki hücrelerinin içinde glukoseramid birikimi meydana gelir ve hastalık bulguları ortaya çıkar.
Gaucher hastalığı GBA1 (Asit-b-glukosidaz) 1q21 genindeki mutasyon sonucu ortaya çıkar. Hastalık anne ve babadan (otozomal resesif) genetik olarak geçiş göstermektedir.
Hastalıkta özellikle dalak, karaciğer, kemik iliği, beyin, daha nadir olarak akciğer, deri, böbrek, ve kalpte glukoseramid birikim görülmektedir.
Buna bağlı olgularda karaciğer, dalak büyümesi kan hücrelerinde düşüşe bağlı kansızlık akyuvar sayısında ve kanamayı önleyen kan pulcuklarında (trombositlerde) azalma saptanır.
Karaciğer veya dalak veya her ikinde birden büyüklük saptanan olgularda mutlaka gaucher hastalığı araştırılmalıdır.
Hastalık damardan verilen enzim tedavisi (enzim yerine koyma tedavisi) ile önlenebilmektedir.
Hastalık klinik bulgularına göre Tip1, 2 ve 3 olarak üç tipe ayrılmaktadır:
En sık görülen klinik formu tip 1’dir.
Tip 2 formu en ağır ve sinir sistemi tutulumunun en ağır olduğu formudur. Bu olgularda yaşam süresi ortalama 9 ay olarak bildirilmektedir.
Tip 1 formunda sinir sistemi bulguların saptanmadığı formudur. Tip 3 ise sinir sistemi bulgularının Tip 2’ye göre daha hafif şekilde görüldüğü görüldüğü formudur.
Tip 3 gaucher hastalarında çocukluk döneminde sinir sistemi bulguların oluşamaması nedeni ile tip 1 tanısı konulabilir. Zamanla sinir sistemi bulgular ortaya çıkabilir. Hastaların bu açıdan takibi önemlidir.
Tip 1 ve 3 hastaları için damar içi enzim yerine koyma tedavisi mevcuttur.
Gaucher hastalığı tiplerini detaylı inceleyecek olursak
Gaucher Hastalığı tip 1’de nörolojik bulgu mevcut değildir. Non-nöronopatik tip olarak bilinmektedir.
Bebeklik dönemden adölesan döneme kadar geniş bir aralıkta hastalık ortaya çıkabilir.
Karaciğer ve dalak büyüklüğü, kansızlık ve kanın pıhtılaşmasında görev alan kan pulcuklarında (trombositlerde) azalma görülmektedir.
Halsizilik, burun kanaması, cildin kolay morarması bulguları görülür.
Dalak büyümesine bağlı ağır karın ağrısı görülebilir.
Olgularda büyüme gelişme geriliği, gecikmiş erkenlik bulguları görülebilir.
Kemik tutulumu, ağır kemik ağrılarına, kemik kırıklarına ve kamburluk veya omurga eğriliklerine neden olabilir.
Gaucher hastalığı tip 1 hastalarında nörolojik tutulum olmakla birlikte gaucher tip 3 kadar ağır değildir.
Gaucher Hastalığı tip 2 ise ağır nörolojik bulgular vardır. Bu olgularda yaşam süresi ortalama 9 ay olarak bildirilmektedir.
Gaucher Hastalığı tip 3 adölesan dönemde ortaya çıkmakla birlikte nörolojik bulgular mevcuttur.
Tanı:
Hastalarda enzim eksikliğinin gösterilmesi ve genetik mutasyonun tahlili ile tanı konulabilmektedir.
Tedavi:
Enzim replasman tedavisi: Tüm hastalarda tedavi verilmeye gerek yoktur. Sağlık Bakanlığının belirlediği klinik bulguları olan olgulara enzim tedavisi verilmektedir.
Enzim replasman tedavisi ile karaciğer ve dalak boyutlarında gerileme görülür. Kan değerlerinde düzelme sağlanır. Enzim tedavisinin sinir sistemine geçişi olmadığı için sinir sistemi bulguları üzerine etkisi azdır.
Ülkemize şu anda tedarik edilen imiglucerase (Cerezyme) ve taligluseraz alfa (Elelyso) tedavisi 15 günde bir damardan verilir.
Substurat redüksiyon tedavisi: Eliglustat ve Miglustat (Zavesca) etken maddeli ilaçlar glukoseramid oluşumunu baskılayıp vücutta birikmesini önleyerek etki ederler.
Kemik iliği tranplantasyonu: Enzim tedavisi bulunmadan önce en etkin tedavi yöntemiydi. Belirgin yan etkileri ve hastayı kaybetme riski olduğu için eski önemini yitirmiştir.
Anne ve babadan genetik olarak geçen metabolik bir hastalıktır. Çekinik genle taşınan bu hastalığın taşıyıcı sıklığı ülkemizde yüksektir. Her 100 kişiden dördünün bu hastalığı taşıyor olması yanı sıra %22 ‘ye varan akraba evliliği sıklığı hastalığın ülkemizde sık görülmesinin nedenidir.
Türkiye fenilketonüri hastalığının en sık görüldüğü ülkeler arasındadır. Doğan her 6.000 – 8.000 çocuktan biri fenilketonüri hastası olarak dünyaya gelmektedir.
Fenilketonüri hastalığının genetik aktarımının şeması:
Ülkemiz hastalığın en sık izlendiği ülkelerdendir. Doğması beklenen bebek sayısı ile değerlendirildiğinde her yıl 200-300 yeni fenilketonüri vakasının topluma katılacağı hesaplanmaktadır.
Hastalıkta protein yapıtaşı olan fenilalanin adlı aminoasidi karaciğerde tirozin adlı aminoaside dönüştüren fenilalanin hidroksilaz enziminin normal çalışmaması sonucu kanda fenilalanin birikimi ile meydana gelir.
Kanda biriken fenilalanin ve geriye dönüşümsüz beyin hasarına neden olur. Erken tanımlanıp tedavi edilmediği takdirde kaçınılmaz son ağır zihinsel geriliktir.
Ağır zihinsel geriliği olan fenilketonürili bireylerde konvülsiyonlar, agresif yada otistik davranış bozuklukları, dermatit şeklindeki cilt lezyonları yanı sıra vakaların % 60’ında anne babaya göre açık saç-göz-ten rengi ile karekterize görünüm vardır.
Fenilketonüri hastalığının kandaki fenilalanin düzeyine göre 2 farklı tipi vardır:
1.Klasik Fenilketonüri: Kanda bakılan fenilalanin düzeyi 20 mg/dl (1200 µmol/L) üzerinde olan hastalar bu gruba girer. Bu hastalarda fenilalanin hidroksilaz enzimi hiç çalışmamaktadır.
Klasik Fenilketonüri hastaları tanı konulduğundan itibaren fenilalaninden kısıtlı diyet yapmaları gerekmektedir.
Diyet tedavisi ömür boyudur.
Fenilalanin tüm proteinli gıdaların içinde bulunmaktadır (kırımızı/beyaz et, süt, peynir, yoğurt, yumurta, ekmek…). Bu hastalar, içinde fenilalanin bulunmayan özel ürünler kullanmaları ve vegan diyet (bitkisel gıda ağırlıklı) gerekmektedir.
Diyete erken yaşta başlamak ve uymak zihinsel etkilenmenin önlenmesinde en önemli faktördür.
Hastaların yaşına ve fenilalanin değerlerine göre belli aralıklarla kan fenilalanin düzeyine bakılmalı, diyetleri metabolizma uzmanları ve diyetisyenler ile ayarlanmalıdır. Kanda bakılan fenilalanin düzeyinin 2-6mg/dl (120-360 µmol/L) arasında tutulması ağır zeka etkilenmesini engellemektedir.
2. Hiperfenilalaninemi: Bu grup kendi içinde iki alt gruba ayrılmaktadır
a. Hafif Hiperfenilalaninemi: Normal diyetle izlemde kan fenilalanin düzeyi 2-10 mg/dl (120-600 µmol/L) arasında seyreder.
Bu hastalarda fenilalanin düzeyi 2-6 mg/dl (120-360 µmol/L) arasında olanlarda diyet tedavisine gerek yoktur. Fakat olası fenilalanin düzeyi yükselmelerine karşı rutin Metabolizma Bölümünde takibi gerekmektedir.
b. Hiperfenilalaninemi: Kan fenilalanin düzeyi 10-20 mg/dl arasında olan hastalardır.
Kan fenilalanin düzeyinin 6mg/dl üzerine çıkması durumunda zeka etkilenebileceği için bu grup hastalar da fenilalaninden kısıtlı diyet yapmaları veya ilaç tedavileri almaları gerekmektedir. Bu gruptaki olgular Klasik Fenilketonürili hastalara göre diyet tedavilerindeki kısıtlamalar genellikle daha hafif olmaktadır.
Fenilketonüri hastalarına diyet tedavisi ne kadar erken başlanırsa zeka etkilenmesi o kadar az olur. Bu sebepten dolayı hastalık ülkemizde topuk kanı taraması programına alınmıştır.
Fenilalanin hidroksilaz enziminin çalışmasında gerekli olan tetrahidrobiyopterin’in (BH4) geri dönüşüm bozukluğuna bağlı ortaya çıkan durum “Malign Fenilketonüri” olarak adlandırılmaktadır.
BH4 sadece Fenilalanin Hidroksilaz enziminin değil ayrıca Tirozin Hidroksilaz ve Triptofan Hidroksilaz enziminin de normal çalışması için gereklidir. Bu enzimlerinde çalışamamasına bağlı olarak sinir sisteminin normal çalışması için gerekli olan seratonin ve dopamin sentezi yapılamaz.
Bu olgularda BH4 yanında L-dopa/carbidopa ve 5-OH-triptofan desteği de yapılması gerekmektedir. Bazı olgular fenilalaninden kısıtlı diyete de ihtiyaç duyarlar. Nörolojik etkilenme bu olgularda klasik fenilketonüriye göre daha ağırdır.
Fenilketonüri Hastalarının İzlemi:
Hasta izlemleri kan fenilalanin düzeyi ile yapılmaktadır.
Bebeklerde daha sıkı takip önerilirken yaş arttıkça takip aralıkları da uzamaktadır. Her ülkenin kendisine göre bir takip planı mevcut olmakla birlikte merkezler arası takip planları değişiklik gösterebilmektedir.
Amaç kan düzeyini bebeklerde 2-6 mg/dl (120-360 µmol/L) arasında tutmak iken
12 yaş üstü hastalarda 2- 10 mg/dl (120-600 µmol/L) arası olması önerilir.
Her ne kadar takip aralıkları ülkeden ülkeye değişse de her hasta kendi içinde değerlendirilmelidir.
Tedavinin hedefi diyet ile yeterli büyümenin sağlanması, kan fenilalanin artışına bağlı oluşan nörolojik etkilenmenin ve beslenme kısıtlamalarına nedeni ile oluşabilecek vitamin, mineral ve eser element eksiklerinin önüne geçilmesini sağlamaktır.
Tedavi ömür boyudur.
Erişkin hastalarda diyete uyumsuzluk sonucu kanda yükselen fenilalanin seviyelerinin dikkat eksikliğine, agresif davranışlara, arka arkaya hızlı şekilde yapılan işlemlerde yavaşlama gibi sinirsel ve psikiyatrik sorunlara yol açtığı ve diyete uyumlu ile kan fenilalanin seviyesi düştüğünde bu bulguların ortadan kalktığı gösterilmiştir.
Fenilketonüri hastalarının hayvansal gıda tüketememelerinden dolayı hastalarda B12 vitamini, folik asit, demir, çinko, bakır, magnezyum, fosfat, selenyum, kalsiyum, esansiyel yağ asitleri (çoklu doymamış yağ asitleri) eksiklikleri görülebilmektedir. Bu durumun önüne geçebilmek için, hastalara vitamin ve mineral desteği verilmeli, vitamin mineral eksiklikler açısından belli aralıklarla tetkik edilmeli ve büyüme ve gelişmeleri takip edilmelidir.
Kemik mineral yoğunluğu, bu olgularda nedeni tam olarak açıklanamayan bir sebepten dolayı kendi yaş gruplarına göre geri olabilmektedir. Fenilketonürili hastalarda kemik erimesi (osteoporoz, osteopeni) yönünden değerlendirilmeli ve kalsiyum D vitamini desteği verilmelidir.
Belli aralıklarla rutin nöropsikiatrik testlerle etkilenmeleri ortaya konulmalıdır.
Diyet tedavisi ömür boyudur. Zamanla diyet tedavisinde esneklikler yapılsa bile yükselen kan fenilalanin düzeyi olgularda nöropsikiatrik etkilenmelere neden olmaktadır.
Fenilketonüri Tedavisinde Yenilikler:
Enzim yerine koyma tedavisi: Fenilalanin liyaz enzimi memeli canlılarda olmayan bitkilerde bulunan bir enzimdir. Fenilalanin’i toksik olmayan maddelere dönüşmesini sağlar.
Bu enzim cilt altına iğne ile verilmesi uygulanmaktadır. Dünyada ve ülkemizde uzun dönem tedavi sonuçları araştırılmaktadır.
Large nötral aminoasit içeren tablet ve toz ürünler: Large nötral aminoasitleri (LNAA) (L-Tyrosine, L-Tryptophan, L-Methionine, L-Threonine, L-Isoleucine, L-Valine, L-Leucine, L-Histidine, L-Lysine, L-Arginine, Phe bir LNAA’dır) içeren bir üründür. Bu aminoasitler kandaki fenilalanin beyne geçişini ve bağırsaklardan fenilalanin emilimini engelleyerek sinir sistemini korumaktadır. İlaç yemeklerle birlikte alınmaktadır.
Özellikle erişkin yaş hastalarda kullanılmaktadır. Yüksek fenilalaninin yol açtığı dikkat eksikliğine, agresif davranışlara, seri yapılan işlemlerde yavaşlama gibi sinirsel ve psikiyatrik durumlara olumlu etkileri olduğu belirtilmektedir.
LNAA içeren ürünler Türk Eczacılar Birliğinden tedarik edilmektedir.
Sapropterin (Kuvan, Diterin): Sapropterin fenilalanin hidroksilaz enziminin çalışmasında yardımcı olan bir maddedir. Fenilalanin hidroksilaz enziminin kısmı eksikliğinde mevcut enzimin dahaz hızlı çalışmasını sağlar. Klasik PKU hastalarının %30’unun ve hafif PKU hastaların %80’inin bu tedaviden fayda gördüğü belirtilmektedir.
Hastaların sapropterine yanıt verip vermeyeceği saptropterin yükleme testi ile yapılmaktadır: sapropterin tablet formu ağızdan verildiğinde kan fenilalanin düzeyinde %30 düşüş saptanması klinik olarak yanıt verdiğini göstermektedir.
Ayrıca genetik tahlil ile de hastanın tedaviden fayda görebileceği belirlenebilir.
Karaciğer nakli: Nakil sonrası oluşabilecek sorunlar düşünüldüğünde önerilmeyen bir yöntemdir.
Gen Tedavisi: Şu an yapılamamaktadır. Klinik çalışmalar devam etmektedir.
Fenilketonürili hastalar için ülkemizde çeşitli dernekler bulunmaktadır.
Bu derneklerde aileler bir araya gelmekte ve çeşitli etkinlikler düzenlenmektedir.
Ayrıca dernek ve Metvak Vakfı aracılığı ile hastaların tüketebileceği özel ürünler tedarik edilebilmektedir.
Karaciğerde bulunan “glisin cleavage” adı verilen enzim kompleksinin doğuştan eksikliği sonucu vücut sıvılarında (kan, beyin-omurilik sıvısı, idarda) glisin amino asidinin birikimi ile seyreden bir metabolik hastalıktır.
Hastalık anne ve babadan genetik (otozomal resesif) olarak geçer.
Bir çok metabolik hastalıkta olduğu gibi Nonketotik hiperglisinemi de taşıyıcı anne ve taşıyıcı babanın sorunlu allellerinin çocuklarında yan yana gelmesi ile oluşur. Taşıyıcı anne babanın çocuklarının hasta olma ihtimali %25 iken %50 ihtimalle çocukları taşıyıcı olur ve %25 sağlam çocuk sahibi olma ihtimalleri vardır. Akraba evlilikleri bu sorunlu allellerin yan yana gelme ihtimalini arttırmaktadır.
Hastaların %85’ten fazlası yaşamın ilk bir kaç gününde bulgu verir. Çok az bir kısmı geç dönemde (ilk bir yıl içinde) bulgu verebilir.
Hastalar doğum sonrası kas zayıflığı, emmede güçlük, solunum durması, bilinç bozukluğu bulguları ile başvururlar. Hıçkırık tarzında nöbetler görülür. Sinir sistemi etkilenmesine bağlı besinleri yutma güçlüğü ve sindirim sistemi hareketlerinin azalmasına bağlı kabızlık şikayetleri görülebilir.
Vücutta biriken glisin sinir sistemine toksik etki yaratır ve buna bağlı olgularda zeka geriliği, nöbet geçirme gibi sorunlar ortaya çıkar. Sinir sistemi bulguları geri dönüşsüzdür.
TANI:
Tanı için beyin-omurilik sıvısında artışının gösterilmesi önemlidir. Ayrıca kanda ve idrarda da glisin miktarı artmıştır.
Beyin MR’da olgularda beyinin bir parçası olan korpus callosum küçülmesi veya yokluğu gibi yapısal bulgular görülebilir.
Beyin elektriksel aktivitesinin izlendiği EEG’de hastalığı özgü bulgular saptanabilir.
Karaciğer biyopsisinde “glisin cleavage” enzim aktivitesinin düşük olduğunun gösterilmesi ile de tanı koymak mümkündür.
Tedavide amaç kan glisin seviyesini toksik seviyelerin altına düşürmektir.
Glisin protein içeren gıdalar içinde bulunduğundan hastalara proteinden kısıtlanmış diyet verilmesi gerekir. Bunun için yapılmış özel glisin içermeyen tıbbi ürünler kullanılabilir.
Sodyum benzoat adlı ilaç ile kandaki glisin düzeyi azaltılabilir. Ayrıca sodyum benzoat nörolojik bulgulara da olumlu etkileri bulunabilir.
Hastalara folik asit vitamini verilebilir.
Ağır nörolojik bulguları ve dirençli nöbetleri olan hastalara Santral sinir sistemine etki eden dekstrometorfan (bexine) adlı ilaç tedavisi verilebilir.
Nöbetlerin kontrolünde antiepileptikler (nöbet ilaçları) ve ketojenik diyet tedavileri diğer seçeneklerdir.
Hastaların kas güçsüzlüğü olması nedeni ile fizik tedavi almaları önerilmektedir.
Ayrıca beslenme ve yutma problemi olan olgular için tüp ile beslenme (PEG) ve reflü engelleyici cerrahi yapılması gerekebilir.
Homogentisat 1,2dioksijenaz enziminin doğuştan eksikliğine bağlı homogentisik asit’in (HGA) maleilasetoasetat’a dönüşememesi sonucu benzoquinon asetik asit (BQA), melanin- benzeri pigmentler birikimi ile seyreden bir hastalıktır.
Homogentisat 1,2 dioksijenaz enzimi geni 3q13.33 kromozunda bulunmaktadır. Hastalık otozomal resesif olarak anneden ve babadan geçer.
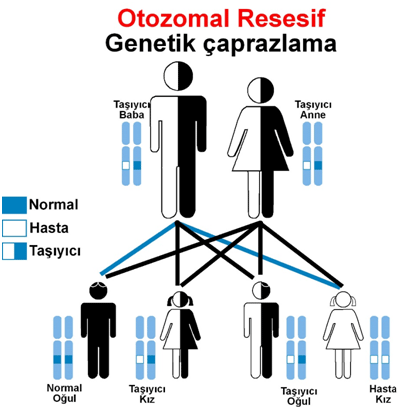
Otozomal resesif geçiş şeması:
Metabolik hastalıkların çoğu otozomal resesif (çekinik kalıtım) şeklinde genetik olarak anne ve babadan hastaya geçmektedir.
Gerekli olan enzimi insan DNA’sında iki adet sağlam allel’den sentez edilmektedir. Eğer bu iki allellerden birinde sorun varda kişi sağlam allel ile gerekli enzimi üretir hasta olmaz fakat hastalığı taşıyıcı olur.
Eğer iki allelde de sorun varda hastalık ortaya çıkar. Bir çok metabolik hastalık gibi Alkaptonüri de taşıyıcı anne ve taşıyıcı babanın sorunlu allellerinin çocuklarında yan yana gelmesi ile oluşur. Taşıyıcı anne babanın çocuklarının hasta olma ihtimali %25 iken %50 ihtimalle çocukları yaşıyıcı olur ve % 25 sağlam çocuk sahibi olma ihtimalleri vardır. Akraba evlilikleri bu sorunlu allellerin yan yana gelme ihtimalini arttırmaktadır.
KLİNİK
Yenidoğan ve bebeklik döneminde bekleyen idrarın siyahlaşması veya bebek bezinde fark edilen siyah renkli idrar çok tipiktir.
İleri yaşlarda şakaklarda kulaklarda ve arkalarında burun üzerinde siyahlık ve göz beyazında siyahlaşma görülebilir.
Adölesan çağda ağırlık taşıyan eklemlerde; omurgada, kalça, diz eklemlerinde madde birikime bağlı eklem problemleri (artropati) ortaya çıkabilir. Eklem ağrısı, hareket kısıtlılığı 30 yaş civarında görülebilir. 20 yaş öncesi nadiren görülür. Kemik mineralizyonunda azlamaya bağlı (osteoporoz) olgularda kemik kırıkları, tendon kopmaları görülebilir. Bazı olgularda böbrek taşı ve tükrük bezi taşı oluşabilir. İşitme kaybı nadirde olsa ilerleyen dönemlerde görülebilir.
Madde birikiminin olduğu önemli bir yer ise kalptir. Buna bağlı kalp yetmezlikleri, kalp kapakçıklarında darlık ve kalp ritim bozuklukları ileri yaşlarda görülebilir.
TANI
Hastalarda idrar homogentisik asit seviyesi aşırı artmış olarak saptanır.
Genetik tahlil kesin tanı konulmasında önemlidir.
Kemik ve eklem bulguları için röntgen ve diğer görüntüleme yöntemleri kullanılabilir.
Klinik bulguların ağırlığının tespiti için çeşitli skorlama sistemleri kullanılmaktadır.
TEDAVİ
Kesin bir tedavisi bulunmamaktadır. Çeşitli tedavi yöntemleri ile homogentisik asit ve benzoquinon asetik asitin birikiminin önüne geçilebilir. Tedavi yöntemlerinin yan etkileri göz önüne alınması gerekmektedir.
Tedavi Seçenekleri:
Askorbik asit (C vitamini): 1000mg/gün 2 dozda Benzoquinon asetik asit oluşumunu antioksidan aktivite ile geriletir. Yüksek doz askorbik asit idrarda Benzoquinon asetik asit atılımını azaltır. Uzun dönem klinik etkinliği tam olarak bilinmemektedir.
Nitisinon tedavisi: Uzun dönem klinik etkinliği tam olarak bilinmemektedir. 4-hidroksi fenilpiruvat dioksijnaz enzimini inhibe ederek biriken maddelerin üretimini durdurarak etki gösterir. Kullanımı sırasında kanda tirozin seviyesi yükselebilir ve tirozinemi adı verilen başka bir metabolik hastalığın bulgularının görülmesine neden olabilir: cilt, göz, sinir sistemi ve karaciğer etkilenmesi açısından takip edilmelidir. Bu yüzden hastalara tirozin ve fenilalaninden kısıtlı diyet tedavileri verilmeli ve diyete uyum sağlanmalıdır. Takibinde plazma homogentisik asit ve tirozin düzeyi takip edilmeli. Tedavi dozu, tedavi başlama yaşı, tedavi yanıtlılığı ve tedavi yan etkileri hakkındaki bir çok soru hala cevap beklemektedir.
Düşük proteinli diyet: Tirozin amino asidinden kısıtlı diyet önerilebilmektedir. Böylece homogentisik asit ve benzoquinon asetik asitin üretimi ve dolayısı ile birikimi önlenebilir. Küçük çocuklarda proteinden kısıtlı diyet büyüme gelişmeyi olumsuz etkilemesi en önemli yan etkisidir.
Yaşam önerileri: Yarışmalı olmayan, eklem ve tendonları zorlamayan sporlar eklem tutulumları ve eklem hareket kısıtlılıklarının önüne geçebilir
Fizyoterapi: Etkilenmiş eklemlerde yaşam kalitesini arttırabilir.
Ağrı kontrolü: Eklem tutulumu olanlarda çeşitli ağrı kesici ilaç ve cerrahi tedaviler kullanılabilir.



































Yorum yazabilmek için oturum açmalısınız.