

Klinik: Metakromatik lökodistrofi (MLD) sinir sistemini tutan bir lizozomal depo hastalığıdır.
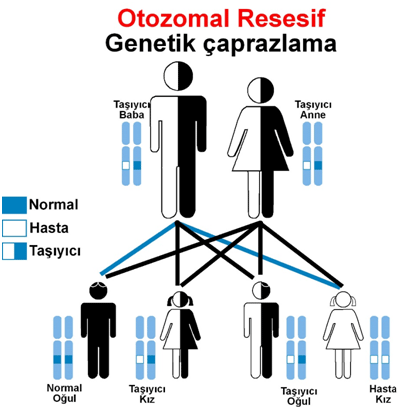
Hastalık hem anneden hem babadan geçmektedir. Otozomal resesif (çekinik) olarak kalıtılmaktadır.
Geç infantil form (Geç bebeklik formu):
En sık görülen formudur.
Bulgular 1-2 yaşında başlar. Olguların çoğu yürüyebilir. %15’i ise yürüyememektedir.
Hastalar ilk bulgu olarak 14-16. Aylarda yürümede ve sıralama güçlük ve düşme şikayetleri ile gelir.
Fizik muayenede kas zayıflığı (hipotoni), periferal sinir sistemi tutulumuna bağlı reflekslerin alınamaması dikkat çeker.
Zamanla ayağa kalkma ve yürüme hastalarda imkansız hale gelir.
Konuşma güçülüğü, zeka geriliği, körlüğe neden olan göz tutulumları ilerleyen dönemlerde bitkisel yaşam ve vefat gerçekleşir.
Juvenil Form (Çocukluk Formu):
3-14 yaş arasında bulgular başlar okul başarısızlığı, davranış problemleri, algılama bozukluğu görülür.
Yürüme bozuklukları, serebellar ataksinin (dengenin sağlamaması) eşlik ettiği sinir sistemi bulguları görülebilir.
Daha nadir olarak nöbet görülebilir.
Adult Form (Erişkin Form):
İki farklı klinik tipte görülür:
1. Motor bulguların ön planda olduğu, denge, kas tonusunun etkilendiği hasta grubu.
2. Davranış ve psikiyatrik bulgular (şizofreni gibi) ön plandadır. Unutkanlık ve sinir sistemi tutulumu ilerleyen dönemde tabloya eklebilir.
Metabolik Yönetim:
Sulfatidlerin ve diğer sülfatlanmış glikolipitlerin lizozom içinde metabolize edilme sorun vardır.
Arilsülfataz-A enzim eksikliğine bağlı görülür.
Sap-B ile arilsülfataz A birlikte çalışarak etki gösterir. Bu yüzden Sap-B eksikliği de MLD tablosuna neden olabilmektedir.
Sulfatidler MLD hastalarında böbreklerde de birikerek idrardan sülfatid atılımına neden olmaktadır.
Genetik:
80’den fazla mutasyon belirtilmiştir.
ARSA geninde 22q13 kromozomda eksprese olur.
459+1G>A: ağır fenotip
P426L: hafif fenotip
I179S: hafif fenotip
Tanı:
Motor sinir iletim hızı azalmıştır.
Erişkin formlarda ise her zaman sinir iletim hızı etkilenmez.
Beyin MR’ı görüntülemede beyaz cevherdeki görünüm önemlidir. (iki taraflı simetrik değişiklikler T2 ağırlıklı görüntülemede diffüz hiperintens alanlar, T1 ağrılıklı görüntülemede hipointens görünüm mevcuttur.)
Beyincik küçülmesi görülmesi sıktır.
Arilsülfataz A enziminin eksikliğinin gösterilmesi ve ARSA gen mutasyonu saptanması tanıda önemlidir.
İdrar sulfatid düzeyi ölçümü tanı açısından önemli bir tetkiktir.
Sap-B eksikliğinde arilsülfataz A düzeyi normal saptanır. Bu hastalarda Genetik olarak PSAP genine bakılmalıdır.
Tedavi:
Hastalığı tamamen ortadan kaldıran bir tedavi yoktur.
Eklem sertliklerinin tedavisi ve ağrılar için radikülopati yapılabilir.
Bazı hafif etkilenmiş hastalarda kemik iliği nakli uygunlanmıştır. Yavaş progresyon gösteren juvenil ve adult formlarda faydalı olabilir. Kemik iliği naklinin sinir sistemi üzerine etkisi kısıtlıdır.
MLD tanı, tedavi ve takibi için merkezimize başvurabilirsiniz. Merkez bilgilerimiz için tıklayınız.
Bu konu hakkında bize soru sorabilirisiniz.

























Yorum yazabilmek için oturum açmalısınız.