
Zellweger Spektrum Bozuklukları (ZSB), peroksizom adı verilen hücre içi organellerin oluşum ve işlev bozukluğuyla karakterize, nadir görülen genetik bir hastalık grubudur. Bu hastalıklar, peroksizomal biyogenez bozuklukları (PBD) olarak da adlandırılır ve genellikle ciddi nörolojik, karaciğer ve diğer organ problemleriyle ilişkilidir.
Hastalığın Temel Özellikleri
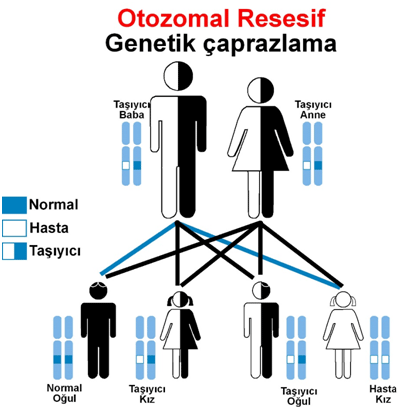
ZSB, genetik geçişli otozomal resesif hastalıklardır. Hastalık anneden ve babadan geçer. Peroksizomlar, hücrelerde uzun zincirli yağ asitlerinin ve diğer toksik maddelerin metabolizmasını sağlayan organellerdir. ZSB’de bu organellerin işlevi bozulduğu için toksik maddeler vücutta birikir.
Zellweger Spektrum Bozukluklarının insidansı yaklaşık olarak 1:50.000 ile 1:100.000 arasında değişir. Hem kadın hem erkekleri eşit oranda etkiler.
ZSB, ağırlıklı olarak PEX genlerindeki mutasyonlardan kaynaklanır ve klinik olarak üç ana formda sınıflandırılır:
- Zellweger Sendromu (ZS): En ağır form.
- Neonatal Adrenolökodistrofi (NALD): Orta şiddetli form.
- Infantile Refsum Hastalığı (IRD): Daha hafif form.
Semptomlar
ZSB’nin klinik belirtileri geniş bir yelpazeye sahiptir ve hastalığın şiddetine göre değişiklik gösterebilir. Genel olarak görülen semptomlar şunlardır:
Nörolojik Belirtiler
- Kas tonusu kaybı (hipotoni)
- Nöbetler
- Gelişim geriliği
- Görme ve işitme kaybı
Karaciğer ve Metabolik Problemler
- Hepatomegali (karaciğer büyümesi)
- Karaciğer fonksiyon bozukluğu
- Kolestaz (safra akımının bozulması)
- Hipoglisemi
Yüz ve İskelet Anomalileri
- Yassı yüz
- Geniş alın
- Yüksek burun köprüsü
- Eklem kontraktürleri (eklem hareket kısıtlılıkları)
Diğer Belirtiler
- Böbrek kistleri
- Gelişme geriliği
- Yenidoğan döneminde hipotermi ve zayıf emme refleksi
Tanı
ZSB’nin tanısı, klinik bulgulara ek olarak laboratuvar testleri ve genetik analizle doğrulanır:
- Kan ve İdrar Testleri
- Uzun zincirli yağ asitlerinin artışı
- Plazma dokosaheksaenoik asit (DHA) seviyesinde değişiklikler
- Plazma fitanik ve pristanik asit artışı
- Genetik Testler
- PEX genlerindeki mutasyonların analizi
- Görüntüleme
- Beyin MR’ında yapısal anomaliler (lissensefali gibi)
- Karaciğer ve böbrek ultrasonu
Tedavi
ZSB’nin spesifik, kesin bir tedavisi bulunmamaktadır. Ancak semptomatik tedavi ve destekleyici bakım hastaların yaşam kalitesini artırabilir. Tedavi yaklaşımları şunlardır:
- Beslenme Desteği
- Fitanik asitten fakir diyet
- Orta zincirli trigliserit (MCT) yağı takviyesi
- Fizyoterapi ve Rehabilitasyon
- Kas tonusunu ve hareket kabiliyetini artırmak için egzersiz programları
- Nöbet Kontrolü
- Antikonvülzan ilaçlarla nöbetlerin yönetimi
- Görme ve İşitme Desteği
- Görme ve işitme cihazları
- Organ Fonksiyonlarının Yönetimi
- Karaciğer ve böbrek komplikasyonlarının izlenmesi ve tedavisi
Yaşam beklentisi
- Zellweger Sendromu gibi ağır formlar genellikle yaşamın ilk birkaç ayında ölümle sonuçlanır.
- Orta ve hafif formlarda hastalar ergenlik veya yetişkinlik dönemine kadar yaşayabilir, ancak yaşam kaliteleri genellikle düşüktür.
Genetik Danışmanlık
ZSB otozomal resesif kalıtımlı olduğundan, her iki ebeveyn de taşıyıcı olmalıdır. Taşıyıcı çiftler için %25 hastalıklı çocuk sahibi olma riski bulunmaktadır. Genetik danışmanlık ve prenatal tanı, gelecekteki gebeliklerde rehberlik sağlayabilir.
Zellweger Spektrum Bozuklukları (ZSB) tanı ve takibi için merkezimize başvurabilirsiniz. Merkez bilgilerimiz için tıklayınız.
Bu konu hakkında bize soru sorabilirisiniz.





















Yorum yazabilmek için oturum açmalısınız.