

STK her birinin başlangıcı ve ciddiyeti farklılık gösteren, hastalığa özgü bir dizi tabloya neden metabolik bir hastalıktır.
STK nadir ve gerçekte olduğundan daha az tanı konulan otozomal resesif bir bozukluktur. Hastalık anne ve babadan otozomal resesif olarak geçer.
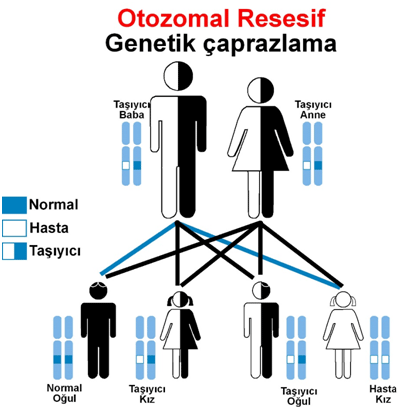
STK’ya mitokondri enzimi olan “27-hidroksilaz”ı kodlayan CYP27A1 genindeki mutasyonlar neden olur.
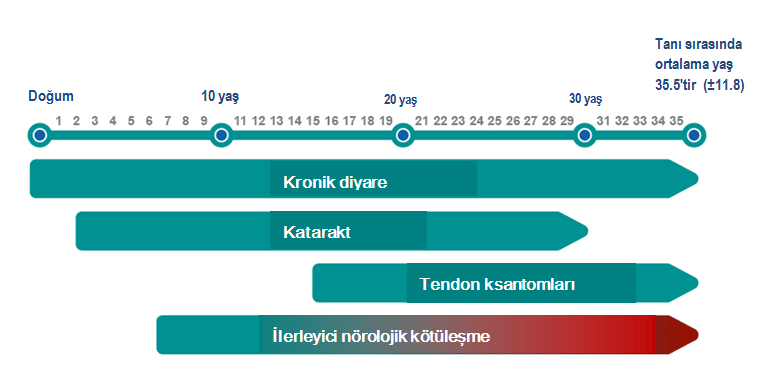
STK’da görülen ayırt ettirici tablolar arasında süt çocukluğunda ya da erken çocuklukta başlayan diyare (ishal), çocukluk çağında ortaya çıkan katarakt, tendon ksantomları ve ilerleyici nörolojik bozulma vardır.

STK’nın değişkenlik gösteren ve birden fazla sistem üzerinde görülen etkileri tanının konulmasında güçlüğe neden olmaktadır.
Bu belirti ve bulguların başlangıçları ve ağırlığı değişkenlik gösterir ve tüm hastalarda hastalığı tanımlayan 4 tablonun hepsi birden olmayabilir.
Ayrıca, hastalığı tanımlayan tablolar farklı organ sistemlerini etkilediğinden, hastaların farklı dallardaki uzmanlara başvurma olasılığı yüksektir ve bu da tanıyı geciktirmekte ve gerçekte olduğundan daha düşük oranda tanı konmasına neden olmaktadır.
STK olgularının yaklaşık yarısında kronik diyare (ishal) geliştiği düşünülmektedir.
Seçili olgu serilerinin sistematik bir incelemesi, STK hastalarında kronik diyare sıklığının %48 olduğunu göstermiştir.
Birçok hasta için diyare en erken bulgudur. Diyare genellikle çocukluk ve süt çocukluğu başlangıçlı olarak tanımlanır, ancak tanı konulmamış hastalarda erişkinlikte de devam edebilir. Tanı konmadan önce hastalar ve onlara bakmakla yükümlü kişiler kronik diyare nedeni ile sık olarak uzmanlara başvururlar.
Tendon ksantomları STK’nın ayrıcı özelliğidir. Tendon ksantomları STK olgularının yarısından fazlasında oluşur. Tendon ksantomları sıklıkla 10-30 yaşları arasında ortaya çıkar ve en sık olarak aşil tendonu etkilenir. STK’da tendon ksantomları, kolestanol, kolesterol ve diğer lipidleri içeren makrofajların tendonları infiltre ederek, bağ dokudaki kollajen liflerini bozmaları ile oluşur. Serum trigliserid ve kolesterol seviyeleri normal olup tendon ksantomu olan tüm hastalarda STK’dan şüphelenilmelidir.
Birçok STK olgusunda açıklanamayan iki taraflı juvenil (sonradan çocukluk çağında ortaya çıkan) katarakt gelişir.
Gençlik döneminde başlayan açıklanamayan kataraktlar STK tanısının konmasında önemli bir fırsattır. STK’da kataraktlar tipik olarak erken başlangıçlı ve iki taraflıdır. Kataraktlar sık olarak 4-18 yaş arasında ortaya çıkar.
Tedavi edilmemiş STK ilerleyici nörolojik sorunlara neden olabilir
İlerlemiş STK hastaları beynin birçok bölgesindeki lipit birikintilerinin ve beyaz cevher kaybının bulgularını gösterirler.
Tanısı atlanan ya da geciken STK Hastaları erişkinlik yaşamlarının ilk yıllarında zihinsel ve fiziksel yetersizlikle yüz yüze kalabilir.
STK’da hastalar genellikle okul çağında geldiklerinde zihinsel yetersizliğin ilk belirtileri vardır. Hastalar yirmili ve otuzlu yaşlarına geldiklerinde zihinsel yetersizlik demansa (unutkanlık) ilerleyebilir.
Erken nörolojik belirti ve bulgular gözden kaçabilir ve kolaylıkla başka sorunlarla karışabilir.
STK hastalarında nöbetler de sık görülür. Tanı konulduğu sırada hastalarda %24 ile %34 arasında değişen oranlarda epilepsi (nöbet) vardır ve başlangıç yaşı hastalar arasında değişkenlik gösterir.
Eğer erken tanı konulup uygun tedavi verilmezse, STK ilerleyerek ciddi fiziksel ve zihinsel yetersizliğe neden olabilir.
TANI:
STK’nın taramasında kullanılan öncelikli biyokimya testi kanda kolestanol testidir.
STK kanda kolestanol seviyesinin yükselmesine neden olur. Normale göre kolestanol seviyelerinin ≥5-10 kat artmış olması yüksek derecede
STK’ya özgü bir durumdur ve hasta bir klinik genetik ya da metabolizma uzmanına yönlendirilmelidir.
Birden fazla sistemi tutma özelliği nedeni ile, STK halen farklı dallardan bir uzmanlar grubu tarafından tedavi edilmektedir.
Genetik analiz ile de tanı konulabilir.
TEDAVİ:
SKT’da vücutta sentezlenemeyen kolik asit ve kenodeoksikolik asit gibi ilaçlar ağız yoluyla hastaya verilerek hastalığın önüne geçilebilir. Erken tanı tedavinin etkinliği özellikle nörolojik etkilerin önüne geçebilmek için önemlidir.
SEREBROTENDİNÖZ KSANTOMATOZİS tanı, tedavi ve takibi için merkezimize başvurabilirsiniz. Merkez bilgilerimiz için tıklayınız.
Bu konu hakkında bize soru sorabilirisiniz.


























Yorum yazabilmek için oturum açmalısınız.