









































Homosistinüri: İdrarda artmış homosistin atılımı anlamında olmakla birlikte esas olarak sistatiyonin β-sentetaz enzim eksikliğine bağlı gerçekleşen ‘’klasik homosistinüri’’yi tanımlamak için kullanılır.
Bu hastalarda doğuştan enzim eksikliğine bağlı olarak vücutta homosisitein düzeyi artar.
Hastalığın dünyada görülme 1:200.000-335000 arasındadır. Akraba evlilikleri tüm metabolik hastalıklarda olduğu gibi bu hastalığında görülme sıklığını arttırmaktadır.
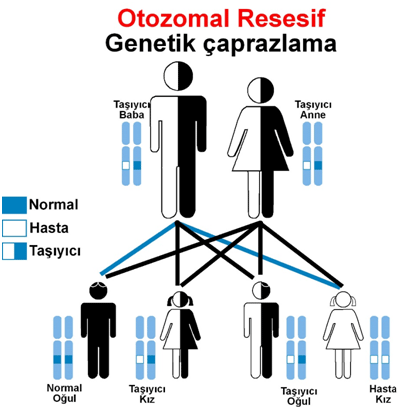
Hastalık anne ve babadan (otozomal resesif) genetik olarak geçer.

KLİNİK:
Gelişme geriliği, entellektüel gerilik: Gelişme geriliği genellikle ilk bulgu olarak görülmektedir.
Tedavisiz olgularda belirgin zeka etkilenmesi olur.
Erken tanı ve tedavi ile zeka etkilenmesi en az seviyeye indirilebilir.
Tedavi edilmeyen olguların %20’sinde nöbetler de görülebilmektedir.
Kişilik bozuklukları, anksiyete, depresyon, obsesif kompulsif bozukluk, psikotik epizotlar görülebilir.
Göz bulguları (Ektopia lentis, ağır miyopi): Miyopi genellikle birinci yılda ortaya çıkarken ektopia lentis (Göz merceğinin normal yerine oranla bir tarafa kayış gösterişi) tedavisiz olgularda ortalama 8 yaşında görülür.
İskelet Bulguları: Kol, bacak kemiklerinde uzama ve uzun boy bu hastalarda sık saptanır.
Olgular osteoporoza (kemik erimesine) yatkındır.
Bunun yanında skolyoz (omurga eğrilikleri), göğüs kemikleri, ayak kemiklerinde eğrilikler görülebilir.
Damar Bulguları: En önemli hayati tehlikeyi oluşturan durumdur.
Artmış homosistein vücut damarlarında hasara yol açar. Bu hasarlı yerlerde kan pıhtılaşması oluşabilir. Oluşan pıhtılaşma inme gibi hayatı tehdit eden durumlara neden olabilir.
Bu hastalarda cerrrahi işlem öncesi vücut sıvı dengesi iyi ayarlanmalı belli anestezikler kullanılmamalıdır. Aksi halde pıhtılaşma riski artabilmektedir.
Tüm klinik bulgular kendi içinde farklılıklar gösterebilir.
TANI:
LAboratuvar tetkikleri ile tanı konulabilmektedir:
1.Kanda artmış homosistin, total homosistein, homosistein-sistein disülfitleri ve metionin düzeyleri artması hastalık tanısı için önemlidir.
2. Azalmış sistatiyonin β-sentetaz enzim aktivitesi gösterilmesi
3.Genetik tanı: 21q22.3 kromozomda kodlanan CBS geninde mutasyon gösterilmesi
TEDAVİ:
Bebeklik döneminde erken tanı koymak ve tedavi başlamak klinik seyri önemli derece olumlu etkilemektedir.
Hastalarda proteinden ve metionin adlı aminoasitten kısıtlı diyet verilmesi gerekmektedir.
Betain tedavisi, B12, folat desteği önemlidir.
B6 vitamin yanıtlı ve B6 vitamini yanıtsız tipleri vardır. B6 vitamini yanıtlı olgularda B6 vitamin tedavisi verilir. B6 vitamini yanıtlı tipin her zaman olmamakla birlikte kliniği daha hafif seyreder.
Tedavinin amacı kanda artmış homosistein seviyesini düşük tutmaktır.
Metionin amino asidi seviyesinin aşırı yükselmesinin önüne geçilmelidir. Tedavide yükselen metionin seviyesi beyin ödemine ve sinir sisteminin olumsuz etkilenmesine neden olabilir. Diyete uyum bu açıdan çok önemlidir.
Homosisteinden sistin amino asidi bu hastalarda sentezlenmediğinden seviyesi normal sınırlarda olmalıdır. Gerekirse suplementasyonu sağlanmalıdır.
Homosistinüri tanı, tedavi ve takibi için merkezimize başvurabilirsiniz. Merkez bilgilerimiz için tıklayınız.
Bu konu hakkında bize soru sorabilirisiniz.




Lizin ve Triptofan amino asitlerinin metabolizması bozukluğudur.
Glutaril-KoA dehidrogenaz adlı verilen enzimin eksikliğine bağlı kan, idrar ve beyin omurilik sıvısında glutarik asit, 3-hidroksiglutrik asit ve glutarilkarnitin birikir.
Biriken maddeler sinir sistemine olumsuz etkileri hastalığın klinik bulgularına neden olmaktadır.
Hastalık anne ve babadan otozomal resesif genetik geçişlidir.
KLİNİK:
Hastaların doğum sonrası büyüme gelişmesi normal seyrederken (6ay- 5yaş) katabolik süreç (vücut metabolizma hızını arttıran durumlar) (aşılama, ateş yüksekliği, enfeksiyonlar gibi) sonrası ani ortaya çıkan (sinir sisitemi hasarlanması) geri dönüşsüz ensefalopati, nöbet, elde edilmiş gelişimlerin kaybı (konuşma ve hareketlerde) meydana gelir. Bu katabolik süreç oluşmadan hastanın tanısını koyup tedavisini başlamak oldukça önemlidir.Etkilenen hastalarda yürüme, beslenme problemleri, nöbet, zeka etkilenmeleri olur.
Bir kısım hasta ise daha nadir olmakla birlikte kas kuvveti zayıflığı (hipotoni), gelişim geriliği, hareket ve duruş bozukluğu bulguları görülebilir. Bu tablodaki hastaya yanlışlıkla diskinetik serebral palsi tanısı konabilir.
Doğumdan itibaren olan baş çevresi büyüklüğü, bu hastalarda görülmektedir. Beyin MR görüntülemede hastalığıa özgü tipik bulgular bu hastalık için oldukça tanı koydurucudur.
TANI:
Tandem: C5 DC (Glutaril) yüksekliği İdrar organik asitlerinde; artmış glutarik asit, 3-hidroksiglutrik asit tanı için önemlidir.
Beyin MR görüntüleme bulguları hastalık için tipiktir.
Genetik analiz (GCDH geni) ile de tanı konulabilir.
TEDAVİ VE KORUMA:
Lizin ve Triptofandan kısıtlı diyet tedavinin temelini oluşturur.
Ayrıca karnitin, riboflavin (B2 vitamini) verilmektedir.
Hayatın özellikle ilk 5 yılında beyin hasarlanmasından korumak esastır. Bu yaştan sonra sinir sistemini ani hasarlanmaya daha dirençli hale gelir.
Akut kötüleşmelerin önüne geçmek için ateş yüksekliği, kusma gibi durumların agresif tedavi edilmesi gerekmektedir.
Nöbet bulguları olan olgulara nöbet ilaçları verilebilmektedir. Kas kasılmaları için fizik tedavi önemlidir. Beslenme, yutma güçlüğü olan olgularda tüp ile beslenme yapılması gerekebilir.
Akut beyin hasarlanması sonrası verilen tedavilerde sinir sistemi bulguları tedavi olmaz.
Glutarik asidüri tip1 tanı, tedavi ve takibi için merkezimize başvurabilirsiniz. Merkez bilgilerimiz için tıklayınız.
Bu konu hakkında bize soru sorabilirisiniz.


Dallı zincirli aminoasit (Valin, Lösin, İzolösin) metabolizmasında kaynaklanan problem sonucu meydana gelir.
Esas bulguların oluşmasında lösin ve metabolitlerinin birikimi sonucu görülür.
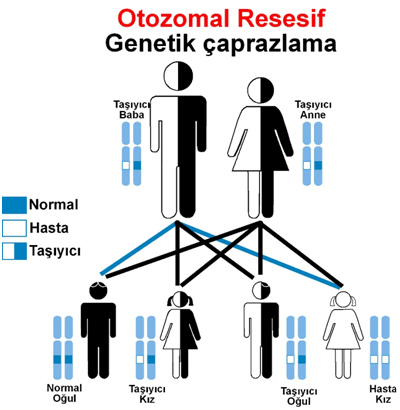
Hastalık anne babadan otozomla resesif olarak kalıtılmaktadır.
Akut tabloda valin, lösin, izolösin yanında artmış organik asitler (a ketoasitler) saptanır
Dallı zincirli a ketoasit dehidrogenaz kompleks aktivitesinde sorun mevcuttur.
Dallı zincirli a ketoasit dehidrogenaz kompleksin E1, E2, E3 komponentleri mevcuttur.
E1: Tiamin pirofosfat bağımlı dekarboksilaz (a ve b komponenti var)
E2: Dihidrolipoil açiltransferans
E3: Dihidrolipoil dehidrogenaz
Bu kompleklerin her birininin sentezindeki sorun hastalığa yol açar.
KLİNİK:
Hastalığın farklı klinik formları mevcuttur.
1. Klasik MSUD:
Yaşamın 1. Haftasında başlayan:
-Uyku hali, beslenmede azalma,
-İlerleyici bilinç kaybı, anormal el kol hareketleri,
-Bulantı, kusma
-Kas tonusunda azalma ve artmalar (hipotoni, hipertoni)
-Nöbet, koma
-İdrarda akçaağaç kokusu (çemen)
Bu hastalarda enzim aktivitesi çok düşüktür.
Tiamin vitamini tedavisine yanıt yoktur.
2. İntermediate MSUD:
Geç başlangıçlı benzer semptomlarla gelirler
Enzim aktivitesi klasik tiplere göre nispeten daha fazladır.
Tiamin vitamini tedavisine yanıt genelde yoktur.
3. İntermitant (aralıklı) MSUD:
Aralıklı denge sorunları, kusma ve ketoasit atakları varıdr.
Enzim aktivitesi %5-20 kadardır.
Ara dönemlerde sağlıklıdır.
Tiamin vitamini tedavisine yanıt genelde yoktur.
4. Tiamin yanıtlı MSUD:
Enzim aktivitesi %2-40
Farmakolojik dozlarda tiamine yanıtı vardır 10 mg/kg/gün (50-300 mg/gün)
5. E3 komponent eksikliği:
Enzim aktivitesi %0-25
Piruvat dehidrogenaz kompleks ve 2-oxoglutarat dehidrogenaz kompleks (mitokondriyal dehidrogenazlar) aktivitesinde de azalma vardır
Yenidoğan döneminde döneminde asidoz ve ilerleyici ensefalopati tablosu mevcuttur.
Diğer hastalardan farklı olarak belirgin Laktik asidoz mevcuttur.
TANI:
Akut tabloda kanda valin, lösin, izolösin yanında artmış a ketoasitler (2-ketoizokaproat, 2-oxo-3-metilvalerat (Akçaağaç kokusunu verir), 2-oxoizovalerat, 2-hidroksiizovalerat, 2-hidroksiizokaproat, 2-hidroksi-3-metilvalerat) saptanır.
Özellikle Lösin artışı hastalarda Lösin ensefalopatisi tablosuna yol açabilir.
E3 komponent patolojilerinde (mitokondriyal dehidrogenazların etkilenmesine bağlı) plazmada laktat, piruvat, alanin artışı; idrarda artmış laktat, piruvat, 2-oxoglutarat, 2-hidroksiizovalerat, 2-hidroksiglutarat
Kanda asidite artmıştır (metabolik asidoz)
Kan ve idrar tahlillerinde keton atılımı mevcuttur.
Kan şekeri düşüklüğü (hipoglisemi) görülebilir.
Genetik analiz (BCKDHA, BCKDHB, DBT) ile de tanı konulabilir.
TEDAVİ:
Kısıtlı lösin, izolösin ve valin alımı ile lösin ve metabolitlerinin oluşumu önlemek
Bir yandan da yeterli miktarda dallı zincirli aminoasit alımı büyüme ve doku onarımı için gerekli
İzolösin ve valin amino asitlerinin yeterli alımı sağlanmalıdır
Amaç büyüme gelişme için yeterli kalori, protein, vitamin ve elementlerin alınması ve normal büyüme ve gelişmeyi devam ettirip metabolit oluşumunu önlemek
Doğal proteinlerden kısıtlı diyet alan olgularda vitamin desteği vermek önemlidir.
Tiamin vitamini yanıtlı olgularda tiamin kullanılabilir.
Akut lösin ensefalopati tablosunda kan lösin değerinin çok arttığı durumlarda diyaliz yapılması gerekebilir.
MSUD tanı, tedavi ve takibi için merkezimize başvurabilirsiniz. Merkez bilgilerimiz için tıklayınız.
Bu konu hakkında bize soru sorabilirisiniz.














Genetik, ilerleyici, geri dönüşümü olmayan, sinir sistemini ve diğer organları tutan bir lizozomal lipid depo hastalığıdır.
Hastalı anne ve babadan genetik olarak geçiş göstermektedir.
Kendine özgü bir fizik muayene, laboratuvar veya MR belirteci olmaması hastalığın tanısının konulmasında zorlaştırmaktadır.
Erken tanı konulması durumunda nörolojik bulguların başında miglustat tedavisi başlanma şansı yakalanabilir. Erken tedavi bulguların önüne geçilmesinde en önemli etkendir.
Hücre içi lipid metabolitlerinin taşınmasından sorumlu (NPC1 NPC2) yapılarda sorun mevcuttur.
Son derece geniş klinik yelpazede karşımıza çıkabilir. Hastalığa özgü olmayan farklı nörolojik ve sistemik bulgular mevcut. Farklı yaşlarda ortaya çıkan semptomlar mevcuttur.
Doğum öncesinden erişkin döneme kadar her dönemde bulgu verebilir
Bulguların şiddeti çok değişkendir. Kardeşlerde bile tablolar çok farklı olabilir. Bu nedenlerle NPC tanısı konulması sıklıkla gecikir.
Tedavinin hangi aşamada başladığı hastalığın klinik şiddeti için çok önemlidir. Mümkün olan en erken dönemde tedaviye başlanmalıdır.
KLİNİK BULGULAR
Uzamış yenidoğan sarılığı, dalak, karaciğer büyümesi, akciğer hastalıkları görülebilir. Kanamayı durduran kan pulcukları (trombosit) sayısında düşüş (trombositopeni) saptanabilir.
Nörolojik bulgular:

TANI
Kesin tanı genetik analiz ile konulmaktadır.
NPC1 (hastaların %95’i) (18q11-q12) (>300 farklı mutasyon var)
NPC2 (hastaların %4’ü) (14q24.3) (18 farklı mutasyon var)
TEDAVİ
Miglustat:
NPC’nin tek etkili tedavi yöntemi substrat redüksiyon tedavisidir
Bu amaçla çocuklarda ve erişkinlerde miglustat (N-butyldeoxynojirimycin) kullanılır.
Nörolojik bulguların başında başlanması tedavi yanıtlılığı için önemlidir.
Hiç nörolojik bulgusu olmayanlarda uzun süre semptom görülmeyebilir
Yaşam süresi nörolojik bulguların şiddeti ve başlama zamanı ile ilişkili
Yutma güçlüğü ve buna bağlı besinlerin akciğere kaçması: En önemli ölüm nedenidir
Miglustat başlamak hasta yaşam süresini anlamlı olarak arttırmaktadır.
Ancak nörolojik bulguları erken başlayanlarda tedavinin etkinlik daha azdır.
Niemann Pick Tip C tanı, tedavi ve takibi için merkezimize başvurabilirsiniz. Merkez bilgilerimiz için tıklayınız.
Bu konu hakkında bize soru sorabilirisiniz.


Tirozin aminotansferaz (TAT) enzim eksikliğine bağlı gelişir.
Kromozom 16q22.1-q22.3 TAT gen mutasyonu bağlı ortaya çıkar.
Hastalık anneden ve babadan otozomal resesif olarak kalıtılır.
Palmoplantar hiperkeratoz denilen el içi ayak tabanında deri bulguları, gözde herpetiform korneal ülserler denilen karneal etkilenme ve zeka etkilenimi gözlenir.
Karaciğer ve böbrek tutulumu olmaz.
Göz bulguları ilk aylarda, cilt bulguları 1 yaş civarında görülür.
Belirgin hipertirozinemi görülür (plazma tirozin seviyesi 500 µmol/L üzerindedir).
İdrarda p-hidroksifenilpiruvat, p-hidroksifenillaktat, p-hidroksiasetat artmıştır. N-asetiltirozin, 4- tiramin de idrar organik asitlerinde artmış olabilir.
Tedavide tirozin ve fenilalanin kısıtlı diyet önerilir.
Tedavi edilmemiş annelerin bebeklerinde, mikrosefali ve gelişim geriliği tanımlanmıştır (maternal tirozinemi).
Tirozinemi tip 2 tanı, tedavi ve takibi için merkezimize başvurabilirsiniz. Merkez bilgilerimiz için tıklayınız.
Bu konu hakkında bize soru sorabilirisiniz.


Krabbe hastalığı merkezi sinir sistemini tutan doğuştan bir metabolik hastalıktır.
Galaktoseramidi (sinir sisteminde bulunan myelin adı verilen sinir kılıfının yapısında yer alan bir madde) hücrelerde bulunan lizozom adı verilen yapılar içinde parçalanmasını sağlayan galaktoseramidaz (galaktoserebrosidaz, serebrozis b-galaktosidaz) enzim eksikliğine bağlı oluşur.
Hastalık otozomal resesif olarak anneden ve babadan geçer.
Galaktoseramidaz geni (GALC) 14q31 kromozomda yer alır. Bu gendeki mutasyonlar hastalığın ortaya çıkmasına neden olur.
Toplumda görülme sıklığı 100.000-200.000 doğumda 1 olduğu belirtilmektedir.
Olguların %85’inden fazlası klasik infantil form denilen bulguların ilk bir yıl içinde başladığı formdur.
Klasik İnfantil Form:
İlk bulgular genellikle ilk 3-6 ayda başlar.
Başlangıç bulguları huzursuzluk, ağlama, kusma, beslenme problemleri, ışığa ve sese karşı kasılmalardır.
Ara ara olan ve nedeni açıklanamayan ateş yüksekliği bu olgularda sık görülür.
Olgularda ilerleyen dönemlerde nöbetler görülebilir.
Ateş yüksekliği ve aşırı salya oluşması da bu hastalarda sık görülür.
Beynin belli bölgelerinin etkilenmesine bağlı kaslarda zayıflık ve körlük meydana gelebilir.
Klasik formlarında hastaların ortalama yaşam süresi genellikle 2 yaştır.
Hastalar genellikle solunumsal problemleri, salya ve yediklerinin soluk borusuna kaçması nedeni ile kaybedilmektedir.
Geç infantil ve juvenil form:
Geç başlangıçlı formunun tanısının konulması klasik hastalara göre çok daha zordur.
15 ay ile 10 yıl arasında bulgular görülmeye başlar.
Çoğu olgu 5 yaşından önce bulgu verir.
İlk bulgu genellikle yürüme bozukluğudur.
Denge problemleri bacaklarda sinirsel sorunlara bağlı yürüme sorunları ortaya çıkar.
Göz sinirlerinin etkilenmesine bağlı görme kaybı bu olgularda sık görülmektedir.
Başlangıç yaşı ve zeka etkilenmesi ve etkilenme derecesi hastadan hastaya değişiklik görülebilir.
Tanı:
Manyetik rezonans görüntülemede (MR) ve Bilgisayarlı tomografide (BT) beyinde etkilenmiş alanların görülmesi hastalığın tanısı açısından önemlidir.
Kanda enzim eksikliğinin gösterilmesi ve genetik mutasyon analizi kesin tanı konulmasında önemlidir.
Tedavi:
Hastalığın kesin tedavisi yoktur.
Destek tedavisi olarak ağrı kesici tedaviler verilebilir.
Kemik iliği veya kord (göbek bağı) kanı nakli bulguların başlamasını önlemede veya hastalığın ilerlemesini önlemede geç başlangıçlı formlarda işe yarabileceği belirtilmektedir.
Ağır bulguları olan bebeklerde kemik iliği naklinin etkinliği zayıf olmakla birlikte anne karnında tanı alan olgularda yenidoğan döneminde kemik iliği nakil yapılmasının (12-44. gün) faydalı olabileceği belirtilmektedir.
Krabbe hastalığı tanı ve takibi için merkezimize başvurabilirsiniz. Merkez bilgilerimiz için tıklayınız.
Bu konu hakkında bize soru sorabilirisiniz.

Yorum yazabilmek için oturum açmalısınız.