




































Klasik galaktozemi, galaktoz-1-fosfat üretemeyen veya işleyemeyen bir enzim olan galaktoz-1-fosfat üridiltransferaz (GALT) eksikliği nedeniyle oluşan kalıtsal bir metabolik bozukluktur. Bu durum, galaktozun normal olarak parçalanmasını ve işlenmesini engeller.
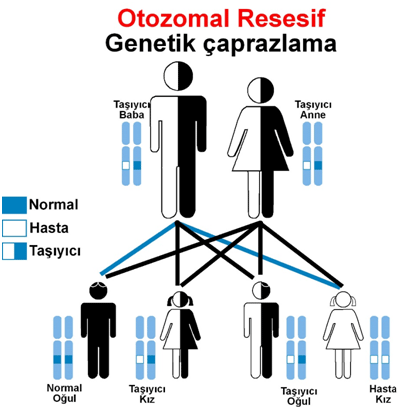
Klasik galaktozemi, otozomal resesif bir kalıtım deseni gösterir. Yani, hastalık için etkili bir genin her iki kopyasını taşıyan bireylerde belirtiler ortaya çıkar. Anne ve babanın her ikisi de taşıyıcı ise, her gebelikte çocuklarının klasik galaktozemi geliştirme olasılığı vardır.
Klasik Galaktozemide Klinik Belirtiler:
Yenidoğan döneminde;
Doğumdan hemen sonra sağlıklı görünen bebeğin anne sütü veya formül mama ile beslenmeye başlamasından birkaç gün sonra:
Beslenme güçlüğü kilo kaybı, kusma – ishal ortaya çıkar.
Sarılık, Karaciğer büyüklüğü karaciğer yetmezliği, Zayıf beslenme- yetersiz büyüme, Böbrek etkilenimi, Katarakt görülebilir.
Laboratuvar bulgularında: İdrarda galaktoz atılımı, kanda sarılık değerlerinde yükseklik, karaciğer testlerinde bozulma görülür.
Yenidoğan döneminden itibaren galaktoz kısıtlaması ile tedavi edilen bebekler, 18-36 aylık döneme kadar sağlıklı bebeklerden ayırt edilemeyebilir, dil ediniminde gecikme ve konuşma problemleri ortaya çıkabilir.
Beyin etkilenimi, Bilişsel bozukluk, öğrenme güçlüğü, okul başarı düşüklüğü, Konuşma, dil ve ses sorunları, Motor bozukluklar, hafıza bozuklukları, sosyal katılım zorlukları, Ortalamanın altında bilişsel süreçler kümesi (dikkat kontrolü, engelleyici kontrol, çalışma belleği, bilişsel esneklik, akıl yürütme, problem çözme ve planlama), Psikiyatrik semptomlar ve duygusal problemler, depresyon, anksiyete, obsesif kompulsif bozukluk ve otizm spektrum bozukluğu, Nörolojik komplikasyonlar (titreme, ataksi, distoni, dismetri ve dizartri) görülebilir.
Klasik galaktozemisi olan kadınlarda gecikmiş puberte, primer amenore, sekonder amenore veya oligomenore ile erken menopoza kadar değişen durum olabileceği, galaktozemili kız hastalarda yumurtalık rezervi için gerekli olan antimüllerian hormonun düşük olduğu, infertilite riskinde ciddi artış olduğu belirtilmiş ancak spontan doğumların da olabileceği rapor edilmiştir.
Erkeklerde testosteron ve inhibin B seviyelerindeki ve sperm sayısındaki hafif azalma, sertoli ve Leydig hücre fonksiyonunda hafif kusurlar olabilir ama ciddi infertiliteye sebep olmaz.
Diğer sistem etkilenimleri
Galaktozemide kemik mineral yoğunluğu normal çocuklara göre daha düşüktür.
Kabızlık ve bulantı normal topluma göre daha fazladır.
Galaktozun diyetle kısıtlanması bu neonatal komplikasyonları çözer ancak erken tanıya ve diyetin erken uygulanmasına rağmen, hastalar beyni ve gonadları etkileyen uzun vadeli komplikasyonlar geliştirmeye devam etmektedir.
Teşhis- Diyet yönetimi
Kırmızı kan hücrelerinde GALT enzim aktivitesi (yok veya önemli ölçüde azalmış) ölçülmeli ve/veya GALT gen analizi ile galaktozemi tanısı doğrulanmalıdır.
Kırmızı kan hücresi GALT enzim aktivitesi %10’un altında olan hastalar ve/veya GALT geninin her iki allelinde patolojik varyasyonları olanlar galaktoz kısıtlı diyetle tedavi etmelidir.
Bir bebekte klasik galaktozemiden şüpheleniliyorsa, hastaya derhal galaktoz kısıtlı bir diyete (örn. Soya bazlı, kazein hidrolizat veya elemental formül), tanının doğrulanmasını beklemeden başlanmalıdır.
Galaktozemili hastaların, süt ürünlerinden yalnızca laktoz ve galaktoz kaynaklarını ortadan kaldıran, yaşam boyu galaktoz kısıtlamalı bir diyetle tedavi edilmesi gerekmektedir.
Klasik galaktozemi diyetinde her tür meyve, sebze, baklagil, fermente edilmemiş soya bazlı ürünler, olgun sert peynirler (galaktoz içeriği <25mg/100g) ve gıda katkı maddeleri sodyum veya kalsiyum kazeinata izin verilir.
Tüm fermente soya bazlı ürünlere tipik olarak diyette kullanılan küçük miktarlarda izin verilebilir (Galaktoz açısından daha yüksek olmasına rağmen).
Hem kalsiyum hem de D vitamini, genel popülasyon için yaşa özel şekilde desteklenmelidir ve yıllık diyet değerlendirilmelidir.
Alınmaması gereken gıda ve ilaçlar:
Anne sütü, laktoz içeren tescilli bebek mamaları, inek sütü, süt ürünleri ve kazein veya peynir altı suyu içeren yiyecekler
Laktoz veya galaktoz içeren ilaç preparatları
Özellikle bebeklik döneminde laktoz içeren ilaçlar (tabletler, kapsüller, tatlandırılmış şuruplar).
Genetik Danışmanlık:
Klasik galaktozemi, otozomal resesif bir hastalıktır. Her ikisi de patojenik GALT mutasyonunun taşıyıcısı olan ebeveynler için, etkilenen bir çocuğa sahip olma şansı her gebelikte %25’tir.
Klasik galaktozemi tanı, tedavi ve takibi için merkezimize başvurabilirsiniz. Merkez bilgilerimiz için tıklayınız.
Bu konu hakkında bize soru sorabilirisiniz.




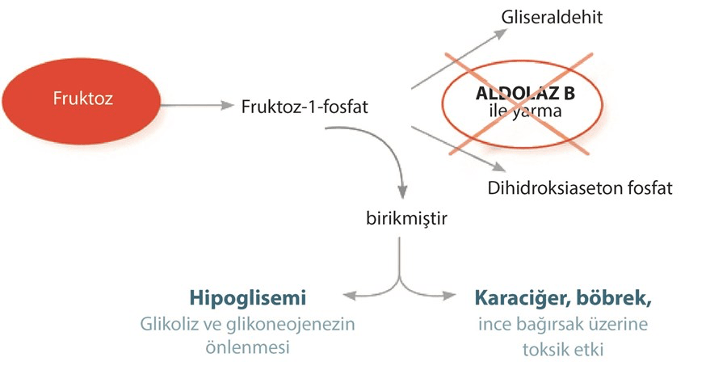
Herediter fruktoz intoleransı (HFI), fruktoz adı verilen bir şekerin işlenmesini engelleyen kalıtsal bir bozukluktur. HFI, fruktoz intoleransı olarak da bilinir ve karaciğerdeki bir enzim olan fruktoz-1-fosfat aldolaz eksikliği nedeniyle ortaya çıkar. Bu eksiklik fruktozun etkili bir şekilde parçalanmasını engeller, fruktoz birikir ve toksik etkilere neden olur.
Herediter fruktoz intoleransı (HFI), otozomal resesif bir kalıtım deseni gösterir. Bu, hastalığın anne ve babadan geçtiği ve bireyin her iki ebeveyninden etkilenen geni taşıması durumunda hastalığın ortaya çıkacağı anlamına gelir.

HFI’ye neden olan genetik mutasyon, fruktoz-1-fosfat aldolaz enziminin etkinliğini etkiler. Bu mutasyon, ALDOB (fruktoz-1-fosfat aldolaz B geni) adı verilen bir genin değişmesiyle oluşur.
HFI’nin belirtileri ve semptomları fruktoz tüketiminin ardından ortaya çıkar. En sık rastlanan belirtiler arasında karın ağrısı, şişkinlik, bulantı, kusma, ishal, zayıflık, halsizlik, solgunluk, hipoglisemi (düşük kan şekeri), büyüme ve gelişme geriliği, sarılık ve karaciğer hasarı yer alır.
Herediter fruktoz intoleransı (HFI) tanısı için aşağıdaki adımlar genellikle izlenir:
Fruktoz Yükleme Testi: Bu testte, bireye belirli bir miktarda fruktoz verilir ve kan şekerindeki değişiklikler izlenir. Normalde, fruktoz karaciğerde parçalanır ve kan şekerine dönüşür. HFI olan bireylerde ise fruktoz işlenemez ve kan şekerinde düşüş görülür.
Fruktoz-1-Fosfat Aldolaz Enzim Aktivite Testi: Bu testte, fruktoz-1-fosfat aldolaz enzim aktivitesi ölçülür. HFI olan bireylerde, bu enzim eksik veya işlevsizdir.
Genetik Testler: Tanı kesinleştirilmezse veya kesin bir teşhis için ihtiyaç duyulursa, genetik testler yapılabilir. Genetik testler, HFI’ye neden olan genetik mutasyonları tespit edebilir.
Tedavi:
HFI tedavisi, fruktoz ve sakkaroz (meyve şekeri) içeren gıdalardan kaçınmayı içerir. Hasta ve ailesi, fruktoz içeriği yüksek olan yiyecekleri ve içecekleri tespit etmek ve bunlardan kaçınmak için diyetlerini dikkatlice düzenlemelidir. Hasta genellikle bir diyetisyen veya beslenme uzmanıyla çalışarak, fruktoz içeriği düşük veya fruktoz içermeyen bir diyet planı oluşturur.
Herediter fruktoz intoleransı olan bir birey, yaşam boyu özel bir diyet uygulamalıdır. Tedavi edilmeyen veya tedaviye uyum göstermeyen durumlarda, ciddi komplikasyonlar ortaya çıkabilir, özellikle karaciğer hasarı ve karaciğer yetmezliği gibi durumlar gelişebilir.
Herediter fruktoz intoleransı tanı, tedavi ve takibi için merkezimize başvurabilirsiniz. Merkez bilgilerimiz için tıklayınız.
Bu konu hakkında bize soru sorabilirisiniz.



Ailesel hiperkolesterolemi (AH), kalıtsal bir lipid metabolizma bozukluğudur ve yüksek kolesterol düzeylerine yol açar. AH’nin temel nedeni, vücutta kolesterolü düzenleyen LDL reseptörlerinde bir bozukluk veya eksiklik olmasıdır. Bu durum LDL kolesterolünün yüksek seviyelerde birikmesine ve arterlerde plak oluşumuna yol açarak kalp hastalığı riskini artırır.
AH, genetik bir bozukluktur ve genellikle otozomal dominant yolla aktarılır. Bu, bir ebeveynin etkilenen bir geni taşıdığında, her çocuklarının bu bozukluğu geliştirme olasılığının yüzde 50 olduğu anlamına gelir. AH olan bir bireyin çocuk sahibi olmadan önce genetik danışma alması önerilir.

Fizik Muayene ve Semptomlar:
Doktorunuz, yüksek kolesterol ile ilişkili semptomları ve fizik muayene bulgularını değerlendirecektir. Örneğin, tendonlarda kolesterol birikimi (ksantoma) veya göz kapaklarında kolesterol birikimi (ksantelazma) gibi belirtiler AH’yı düşündürebilir.
Erken Tanı ve Tarama:
AH genellikle semptomlar olmadan ilerleyebilir, bu nedenle erken tanı önemlidir. Aile üyelerinin kolesterol düzeylerini düzenli olarak kontrol ettirmek, erken teşhisi mümkün kılar ve tedaviye başlama fırsatı verir.
Kan Testleri: AH tanısının kesinleştirilmesi için kan testleri yapılır. Tipik olarak, total kolesterol, LDL kolesterol, HDL kolesterol ve trigliserid seviyelerini ölçmek için lipid profili testi yapılır. AH tanısı için LDL kolesterol düzeyleri önemlidir. Yüksek LDL kolesterol düzeyleri, tanıda belirleyici bir faktördür.
Genetik Testler: AH tanısı kesinleştirilmezse veya kesin bir teşhis için ihtiyaç duyulursa, genetik testler yapılabilir. Bu testler, LDL reseptör genindeki veya diğer ilgili genlerdeki mutasyonları tespit edebilir. Genetik testler, ailedeki diğer bireylerin taşıyıcı olup olmadığını belirlemek için de kullanılabilir.
TEDAVİ:
Diyet ve Yaşam Tarzı Değişiklikleri:
AH olan bireylerin sağlıklı bir diyet benimsemeleri ve yaşam tarzı değişiklikleri yapmaları önemlidir. Doymuş yağ ve trans yağ alımını sınırlamak, lifli gıdaları tercih etmek, düzenli egzersiz yapmak ve ideal vücut ağırlığını korumak, kolesterol düzeylerini kontrol altında tutmaya yardımcı olabilir.
İlaç Tedavisi:
Diyet ve yaşam tarzı değişikliklerine rağmen, kolesterol düzeyleri hala yüksek olan bireyler için ilaç tedavisi düşünülebilir. Statinler, kolesterol sentezini azaltarak LDL kolesterol düzeylerini kontrol etmede etkili olabilir. İlaç tedavisi, bir kardiyolog veya uzman doktor tarafından yönlendirilmelidir.
Ailesel hiperkolesterolemi tanı, tedavi ve takibi için merkezimize başvurabilirsiniz. Merkez bilgilerimiz için tıklayınız.
Bu konu hakkında bize soru sorabilirisiniz.


Alfa Mannosidoz, alfa-mannosidaz enziminin eksikliği ile karakterize nadir görülen bir genetik hastalıktır. Bu durum kompleks şeker moleküllerinin çeşitli organ ve dokularda birikmesine yol açarak çok çeşitli semptomlara yol açar.
Alfa Mannosidoz, MAN2B1 genindeki mutasyonların neden olduğu otozomal resesif bir hastalıktır. Bir çocuğun etkilenmesi için her iki ebeveyn de mutasyona uğramış genin bir kopyasını taşımalıdır.
Klinik Özellikler
Alfa Mannosidoz, gelişimsel gecikmeler, zihinsel yetersizlik, iskelet anormallikleri, işitme kaybı, tekrarlayan enfeksiyonlar, yüzde kabalaşma ve organomegali (genişlemiş organlar) dahil olmak üzere bir dizi semptomla kendini gösterir.
1.Zihinsel Yetersizlik ve Gelişimsel Gecikmeler:
Alfa Mannosidozlu çocuklar genellikle çeşitli derecelerde zihinsel yetersizlik yaşarlar. Gecikmiş konuşma ve motor beceriler de dahil olmak üzere gelişimsel dönüm noktalarını geciktirmiş olabilirler.
2.Yüz Kabalaşması ve İskelet Anormallikleri:
Zamanla, Alfa Mannosidozlu bireyler belirgin bir alın, kaba yüz özellikleri, geniş bir burun ve büyük bir dil gibi belirgin yüz özellikleri geliştirebilir. Genişlemiş eklemler, kalınlaşmış kemikler ve boy kısalığı gibi iskelet anormallikleri de mevcut olabilir.
3.İşitme kaybı:
İşitme bozukluğu, Alfa Mannosidozun ortak bir özelliğidir. Hafif ila şiddetli arasında değişebilir ve gecikmiş konuşma gelişimine neden olabilir.
4.Tekrarlayan Enfeksiyonlar:
Bozulmuş bağışıklık fonksiyonu nedeniyle, Alfa Mannosidozlu bireyler, özellikle solunum ve idrar yollarında sık sık enfeksiyonlar yaşayabilir. Bu tekrarlayan enfeksiyonlar, doğada bakteriyel, viral veya mantar olabilir.
5.Organomegali:
Organomegali olarak bilinen genişlemiş organlar, Alfa Mannosidozda ortaya çıkabilir. Karaciğer ve dalak en sık etkilenir ve hepatomegali (genişlemiş karaciğer) ve splenomegali (genişlemiş dalak) ile sonuçlanır.
6.Nörolojik Belirtiler:
Alfa Mannosidoz ilerledikçe, nörolojik semptomlar daha belirgin hale gelebilir. Bunlar arasında ataksi (bozulmuş koordinasyon), kas zayıflığı, titreme, nöbetler ve denge ve yürüme ile ilgili sorunlar yer alabilir.
7.Görüş problemleri:
Alfa Mannosidozlu bazı bireyler, görme keskinliğini etkileyebilecek korneanın bulanıklaşması (kornea opaklığı) veya retina dejenerasyonu gibi görme sorunları geliştirebilir.
Teşhis ve Hastalık Yönetimi:
Alfa Mannosidozun teşhisi klinik değerlendirme, enzim aktivite testi ve genetik testi içerir. Erken teşhis, zamanında müdahale ve destek için çok önemlidir.
Hematopoietik kök hücre nakli (HSCT), alfa-mannosidaz enzim eksikliğinden kaynaklanan nadir bir genetik bozukluk olan Alfa Mannosidozu olan kişiler için potansiyel bir tedavi seçeneğidir. Kemik iliği nakli olarak da bilinen HSCT, kusurlu veya eksik hücrelerin yerine sağlıklı kök hücrelerin hastaya verilmesini içerir.
HSCT, eksik enzimi üreten bir hücre kaynağı sağlayarak Alfa Mannosidozun altında yatan nedeni kontrol altına alma potansiyeli sunar. Uyumlu bir vericiden (allojenik transplantasyon) veya hastanın kendisinden (otolog transplantasyon) elde edilebilen nakledilen kök hücreler, kemik iliğine göç etme ve alfa üretmekten sorumlu olanlar da dahil olmak üzere çeşitli kan hücresi tiplerine farklılaşma kapasitesine sahiptir.
Bireysel bir hasta için HSCT’nin uygunluğunu ve potansiyel faydalarını belirlemek için HSCT’de ve Alfa Mannosidozun yönetiminde deneyimli uzman bir tıbbi ekibe danışmak çok önemlidir. Ayrıntılı bilgi sağlayacak, riskleri ve olası sonuçları tartışacak ve karar verme sürecine rehberlik edeceklerdir.
HSCT’nin Alfa Mannosidozlu bireylerde mevcut nörolojik hasarı tam olarak tersine çeviremeyebileceğini ve etkinliğinin kişiden kişiye değişebileceğini akılda tutmak önemlidir.
Velmanase alfa, biyoteknoloji ile üretilen bir enzimdir. Velmanase alfa, eksik veya kusurlu alfa-mannosidaz enzimini yerine geçerek çalışır. Bu ilacı intravenöz olarak uygulayarak vücuda eksik olan enzimi sağlar ve birikmiş kompleks şekerlerin parçalanmasını kolaylaştırır.
Velmanase alfa ile tedavi, semptomları hafifletmeyi ve Alpha Mannosidozun ilerlemesini yavaşlatmayı amaçlar. İskelet anormallikleri, organomegali (büyümüş organlar) ve genel yaşam kalitesi gibi durumun belirli yönlerini iyileştirmeye veya stabilize etmeye yardımcı olabilir. Bununla birlikte, velmanase alfa’nın mevcut nörolojik hasarı tersine çevirmeyebileceğini veya Alpha Mannosidoz ile ilişkili tüm semptomları tamamen ortadan kaldıramayacağını not etmek önemlidir.
Tıbbi Bakım: Düzenli tıbbi takipler, durumun ilerlemesini izlemek için çok önemlidir. Tedavi, semptom yönetimini, kemik iliği nakli veya enzim replasman tedavisini ve spesifik komplikasyonları ele almak için destekleyici bakımı içerebilir.
Eğitim Desteği ve Gelişimsel Müdahaleler:
Erken Müdahale: Konuşma terapisi, mesleki terapi ve fizik tedaviyi içeren erken müdahale programları, çocuğun genel gelişimini destekleyebilir ve fonksiyonel yeteneklerini geliştirebilir.
Özel Eğitim: Bireyselleştirilmiş bir eğitim planı geliştirmek için özel eğitim uzmanlarıyla işbirliği yapmak, akademik ve sosyal ihtiyaçlar için özel olarak hazırlanmış destek sağlayabilir.
Sağlıklı Yaşam Tarzı: Besleyici diyet seçenekleri, düzenli fiziksel aktivite (uygun olduğu şekilde) ile sağlıklı bir yaşam tarzını teşvik etmek, genel refah için çok önemlidir.
Çevresel Değişiklikler: Güvenli ve erişilebilir bir ev ortamı oluşturmak, potansiyel tehlikeleri en aza indirmek ve Alfa Mannosidozlu bireyler için optimum hareketliliği sağlamak için önemlidir.
Alfa mannosidoz tanı, tedavi ve takibi için merkezimize başvurabilirsiniz. Merkez bilgilerimiz için tıklayınız.
Bu konu hakkında bize soru sorabilirisiniz.


Hastalık genetik geçiş göstermektedir ve ömür boyudur. Tedavi de ömür boyudur.
Hastalık öncelikle biotinidaz enzim aktvitesi bakılarak teşhis edilir. Ülkemizde de enzim aktivitesi bakılarak yenidoğanlarda taranmaktadır. Kesin tanı için genetik analiz yapılmalıdır.
Hayır. Hastalık hem anneden hem de babadan genetik olarak geçer. Gebelikte yaşanılan sorunlar biotinidaz eksikliğine neden olmaz.
Evet. Biotinidaz enzim aktivitesi bir çok faktörden (numunenin uygun saklanmaması, yanlış laboratuvar tekniği, sarılık, kullanılan antibiyotikler) etkilenmektedir. Enzim aktivitesi bu faktörlerden dolayı değişken gelebilir.
Hayır. Biotin suda çözünen bir vitamindir. Yani fazlası idrarla dışarı atılır.
Hayır. Biotin tedavisinin amacı vücutta biotinidaz enziminin çalışmaması sonucu meydana gelen biotin vitamini eksikliğinin yerine konmasıdır. Enzim aktivitesine hiçbir etkisi yoktur.
Hayır. Hastalar biotin tedavilerini düzenli kullandıkça ve takiplerini aksatmadıkça diğer sağlıklı kişiler gibi istediklerini yapabilir, yiyebilir, her türlü tedaviyi rahatlıkla alabilir. Sadece enfeksiyon veya ameliyat gibi metabolizma hızını arttıran durumlar süresince biotin tedavisi dozunu arttırmaları biotin vitamini eksikliğinin önüne geçilmesi için faydalı olabilir.
Evet. Hastalar biotin tedavisi alsalar dahi olası nörolojik bulgular açısından aralıklarla takip edilmeli nörolojik muayene, gelişimsel değerlendirme, göz ve işitme muayeneleri yapılmalıdır. Elde edilen muayene bulgularına göre tedavi dozu düzenlenebilir.
Evet. Biotin tedavisi altında bazı laboratuvar yöntemleriyle bakılan 25-OH-D vitamini, parathormon düzeyi, tiroid fonksiyon testleri gibi homon tetkiklerinde yanlış düşüklük veya yanlış yükseklik çıkabilmektedir. Hekimlerin ve hastaların bu açıdan farkındalığının olması yanlış tetkik sonuçları ve yanlış tedavilerin önüne geçilmesi için oldukça önemlidir.
Evet. Yenidoğan taraması ile tanı konulan vakalarda aile bireylerinin (anne, baba, kardeşler) biotinidaz eksikliği açısından taranması konusu oldukça önemlidir. Klinik deneyimimiz ülkemizde tarama programının yeni olması ve tarama programı öncesi doğan semptomsuz vakaların aile içinde rastlanılabileceğini göstermektedir. Bu yüzden tarama ile tanı alan vakaların aile taramasının yapılmasının ve tanı alanların semptomları olmasa dahi tedavi başlanmasının önemli olduğu görülmektedir.
Biotinidaz eksikliği tanı, tedavi ve takibi için merkezimize başvurabilirsiniz. Merkez bilgilerimiz için tıklayınız.
Bu konu hakkında bize soru sorabilirisiniz.

3 farklı tipi mevcuttur.
Tay-Sachs hastalığı (B varyant): HEXA geni 15. kromozomdaki mutasyona bağlı.
Sandhoff hastalığı (0 varyant): HEXB genindeki mutasyona bağlı.
GM2 aktivatör protein defekti (AB varyant): GM2 aktivatör protein genindeki (5.kromozomda) mutasyona bağlı ortaya çıkar.
Hastalık hem anneden hem babadan otozomal resesif (çekinik) kalıtılır.
TAY-SACHS
beta-heksozaminidaz alfa- subuniti mutasyonu sonucu Heksozaminidaz A (alfa-beta heterodimer)
SANDHOFF
beta*heksozaminidaz beta- subuniti mutasyonu sonucu Heksozaminidaz A ve B (beta beta homodimer) görülür.
Infantil (bebeklik) form en sık görülen formudur.
Klinik: Bulguların görülme yaşı ile sınıflandırılmaktadır.
İnfantil Form: Olgularda 4-6 aylarda kuvvet kaybı ve kas zayıflığı en erken bulgusudur.
Tipik sese karşı (hiperakuzi) aşırı reaksiyon irkilmek görülür.
Tipik olarak göz muayenesinde ‘’kiraz lekesi’’ görülür.
İlerleyen dönemlerde olgularda körlük oluşur.
İlerleyen dönemlerde eklemlerde sertlik hareket kısıtlılığı, yutma güçlüğü ve nöbetler görülebilir.
Baş büyüklüğü (makrosefali) saptanabilir.
Hastal arın sinir sisteminin aşırı etkilenmesi nedeni ile genellikle vefatlar gıdaların solunum yoluna kaçması (aspirasyon pnömonisine) bağlı gelişir.
Sandhoff hastalığında bu bulguların yanında diğer organlarda etkilenme ile organomegali ve kemik bulguları nadir de olsa görülür.
Geç İnfant (bebeklik) ve Juvenil (çocukluk) Form (B1 Varyant):
Çoğunlukla hekzosaminidaz A enzim eksikliğine bağlı görülür.
2-10 yaşından itibaren bulgular başlar.
Denge, konuşma problemleri, psikomotor kötüleşeme, eklem sertliği ve hareket kısıtlığı ve nöbet izler.
Gözdeki kiraz lekesi bulgusu genellikle görülmez.
Kronik Adult (erişkin) Form:
Farklı bulgularla kendini gösterir.
Denge kas tonusu bozuklukları, kas güçsüzlükleri, psikozis (olguların %30-50’sinde), tekrarlayan depresyon, göz hereketlerinde sorun ile birlikte seyreden bulgular görülür.
Metabolik Yönetim:
GM2 gangliosidoz katabolizması için GM2 aktivatör protein gerektirmektedir.
Hekzosaminidaz A (alfa-betaheterodimer) GM2 ye bağlanması için GM2 aktivatör proteine ihtiyacı vardır.
Heksozaminidaz B ise hekzosamin içeren diğer diğer glikolipitleri ve glikoproteinleri metabolize eder fakat GM2 gangliosidozu hidrolize etmez.
Tay-Sahcs’da alfa- subuniti defektine bağlı sadece Hekzosaaminidaz A defekti vardır.
Sandhoff’da ise beta- subuniti mutasyonu sonucu Heksozaminidaz A ve B defekti vardır.
GM2 protein aktivatör eksikliğinde enzimler normal işlev görür.
Tüm tiplerde nöronlarda GM2 ganliozid madde birikimi görülür.
GM2 birikimi bebeklik formlarında çok fazla olmakla birlikte çocukluk ve erişkin formlarda nadir görülür.
Sandhoff olgularında ise karaciğerde ve diğer organlarda da birkimi görülür.
Genetik:
HEXA geni, HEXB geninde, GM2 aktivatör protein geninde (5.kromozom)
Tanı:
Tay-Sachs ve Sandhoff hastalığı ayırıcı tanısı lökosit ve fibroblast kültürlerinde enzim tayini ile yapılmaktadır.
Kesin tanı genetik ile konulmaktadır.
GM2 protein aktivatörü eksikliğinde invitro hekzosaminidaz A aktivitesi normaldir.
MRG’da periventriküler beyaz cevherde minimal T2 sinyal artışı görülebilir.
Tedavi:
Kesin bir tedavisi yoktur.
Konvulzyon tedavisi standart tedavi edilmelidir.
Geç başlangıçlı Tay-Sachs formlarında miglustat tedavisi verilebilir. Etkinliği net değildir.
Gen terapisi deneme aşamasındadır rutin kullanımı henüz yoktur.
Tay Sachs ve Sandhoff tanı, tedavi ve takibi için merkezimize başvurabilirsiniz. Merkez bilgilerimiz için tıklayınız.
Bu konu hakkında bize soru sorabilirisiniz.

Bir tür lizozomal depo hastalığıdır.
Hücre içindeki lizozom adı verilen organellerin içinde çeşitli maddelerin metabolize edilmemesi ve birikmesi sonucu ortaya çıkar.
Otozomal resesif (çekinik) olarak kalıtılır. Hastalık hem anneden hem babadan geçiş gösterir.
Hastalık bulgularının görülme yaşına göre klinik olarak farklılık göstermektedir.
Tipik Erken İnfantil Bbebeklik) Form (Tip1): (enzim aktivitesi %0.1)
Yaşamın ilk bir haftasında bebeklerde kas gücü zayıftır (hipotoniktir).
Hastaların baş kontrolü yoktur. 3-6. Aydan itibaren sinir sistemi gelişimi tamamen durur.
Beslenme güçlüğü ve büyüme gelişme geriliği görülmektedir.
Çoğu bebekte kaba yüz görünümü ve ödem mevcuttur.
Bunun yanında büyük dil, kaba diş etleri, burun kökü basıklığı mevcuttur.
Karaciğer ve dalak büyüklüğü her zaman mevcuttur.
Omurga kemiklerinde eğrilik (skolyoz, kifoz) sık görülür.
Yaşamın bir kaç ay sonrasında göz bebeklerinde titreme (nistagmus) sıklıkla görülür.
%50 olguda göz muayenesinde göz arkasında (retinada) kiraz lekesi görülmektedir.
Zamanla kas gevşekliği yerini eklem sertliğine (spastisite) bırakır.
1 yılın sonunda olgular hızlı nörolojik kötüleşme, konöbet, yutma sorunları ve2 yaş öncesinde de vefat görülür.
Geç infantil (bebeklik) form (tip 2):
12-18.ayda bulgular başlar.
Yürümede güçlük oturma ve ayakta durmakta güçlükle kendini gösterir.
Hızlı ilerleyen ağır el ve bacaklarda kuvvet kaybı görülebilir.
Erken infantil tiplerinde görülen kaba yüz görünümleri (dismorfik bulgular) ve karaciğer büyüklüğü genellikle görülmez.
Görme genellikle etkilenmez.
Kronik geç form (tip 3): (enzim aktivitesi %10)
Çocukluk adölesan veya erişkin dönemde görülebilir.
Konuşma problemleri ve kas tonus problemleri en sık bulgulardır.
Bilişsel bozukluk görülebilir.
Göz bulguları yoktur.
Kemik bulguları beklenmez. Hastalığın ilerleyişi çok yavaştır.
Metabolik Yönetim:
GM-1 gangliosidoz Lizozomal asid -galaktosidaz eksikliği ile meydana gelir.
GM1 gangliosidoz hastalarında dokularda glikoprotein derişye-oligosakaridler, GM1 gangliozid ve keratan sülfat maddeleri birikmektedir.
GM1’de esas birikimi beyin dokusundadır.
İlerleyen dönemlerde böbrek etkilenimi bu hastalarda görülenilmekte, idrarda protein atılımı buna bağlı ödem ve vücutta sıvı birikimi olabilmektedir.
Genetik:
GLB1 (3p21.33) 50’den fazla mutasyon bildirilmiştir.
Fenotip-genotip ilişkisi bildirilmemiştir.
Tanı:
Periferik yaymada lenfositlerde vokuolizasyon kemik iliğinde köpüksü histiyositler görülür.
İdrarda muopolisakkaris analizinde keratan sülfat atılımı artmıştır.
Esas tanı -galaktosidaz enzim düzeyi tayini ile konur.
Tedavi ve Klinik takip:
Özel tedavi maalesef yoktur.
Substurat redüksiyon tedavisi (miglustat) özellikle adult onset formlarda denenebilir, fakat etkinliği belirgin değildir.
Hastalarda görülen nöbetler için nöbet ilaçları kullanılabilmekte, yatağa bağımlı ve hareket kısıtlılığı nedeni ile fizik tedavi almaları gerekmektedir.
Hastalar maalesef ilerleyen zamanlarda solunum cihazı ile takip edilmekte ve beslenme sorunları nedeni ile tüp ile beslenmeye (Nazogastrik sonda, Perkütan gastrostomi) geçilmektedir.
GM1 gangliosidoz tanı, tedavi ve takibi için merkezimize başvurabilirsiniz. Merkez bilgilerimiz için tıklayınız.
Bu konu hakkında bize soru sorabilirisiniz.

Klinik: Metakromatik lökodistrofi (MLD) sinir sistemini tutan bir lizozomal depo hastalığıdır.
Hastalık hem anneden hem babadan geçmektedir. Otozomal resesif (çekinik) olarak kalıtılmaktadır.
Geç infantil form (Geç bebeklik formu):
En sık görülen formudur.
Bulgular 1-2 yaşında başlar. Olguların çoğu yürüyebilir. %15’i ise yürüyememektedir.
Hastalar ilk bulgu olarak 14-16. Aylarda yürümede ve sıralama güçlük ve düşme şikayetleri ile gelir.
Fizik muayenede kas zayıflığı (hipotoni), periferal sinir sistemi tutulumuna bağlı reflekslerin alınamaması dikkat çeker.
Zamanla ayağa kalkma ve yürüme hastalarda imkansız hale gelir.
Konuşma güçülüğü, zeka geriliği, körlüğe neden olan göz tutulumları ilerleyen dönemlerde bitkisel yaşam ve vefat gerçekleşir.
Juvenil Form (Çocukluk Formu):
3-14 yaş arasında bulgular başlar okul başarısızlığı, davranış problemleri, algılama bozukluğu görülür.
Yürüme bozuklukları, serebellar ataksinin (dengenin sağlamaması) eşlik ettiği sinir sistemi bulguları görülebilir.
Daha nadir olarak nöbet görülebilir.
Adult Form (Erişkin Form):
İki farklı klinik tipte görülür:
1. Motor bulguların ön planda olduğu, denge, kas tonusunun etkilendiği hasta grubu.
2. Davranış ve psikiyatrik bulgular (şizofreni gibi) ön plandadır. Unutkanlık ve sinir sistemi tutulumu ilerleyen dönemde tabloya eklebilir.
Metabolik Yönetim:
Sulfatidlerin ve diğer sülfatlanmış glikolipitlerin lizozom içinde metabolize edilme sorun vardır.
Arilsülfataz-A enzim eksikliğine bağlı görülür.
Sap-B ile arilsülfataz A birlikte çalışarak etki gösterir. Bu yüzden Sap-B eksikliği de MLD tablosuna neden olabilmektedir.
Sulfatidler MLD hastalarında böbreklerde de birikerek idrardan sülfatid atılımına neden olmaktadır.
Genetik:
80’den fazla mutasyon belirtilmiştir.
ARSA geninde 22q13 kromozomda eksprese olur.
459+1G>A: ağır fenotip
P426L: hafif fenotip
I179S: hafif fenotip
Tanı:
Motor sinir iletim hızı azalmıştır.
Erişkin formlarda ise her zaman sinir iletim hızı etkilenmez.
Beyin MR’ı görüntülemede beyaz cevherdeki görünüm önemlidir. (iki taraflı simetrik değişiklikler T2 ağırlıklı görüntülemede diffüz hiperintens alanlar, T1 ağrılıklı görüntülemede hipointens görünüm mevcuttur.)
Beyincik küçülmesi görülmesi sıktır.
Arilsülfataz A enziminin eksikliğinin gösterilmesi ve ARSA gen mutasyonu saptanması tanıda önemlidir.
İdrar sulfatid düzeyi ölçümü tanı açısından önemli bir tetkiktir.
Sap-B eksikliğinde arilsülfataz A düzeyi normal saptanır. Bu hastalarda Genetik olarak PSAP genine bakılmalıdır.
Tedavi:
Hastalığı tamamen ortadan kaldıran bir tedavi yoktur.
Eklem sertliklerinin tedavisi ve ağrılar için radikülopati yapılabilir.
Bazı hafif etkilenmiş hastalarda kemik iliği nakli uygunlanmıştır. Yavaş progresyon gösteren juvenil ve adult formlarda faydalı olabilir. Kemik iliği naklinin sinir sistemi üzerine etkisi kısıtlıdır.
MLD tanı, tedavi ve takibi için merkezimize başvurabilirsiniz. Merkez bilgilerimiz için tıklayınız.
Bu konu hakkında bize soru sorabilirisiniz.

Yorum yazabilmek için oturum açmalısınız.